| A 29 year-old Woman with a Mass in the Thigh. June, 2003, Case 306-3. Home Page |
Adeboye O. Osunkoya, M.D. and Ravindranauth N. Sawh, M.B., B.S., D.M. (Path) First Published: June 30, 2003.
Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Kar-Ming Fung, M.D., Ph.D. Last update: April 20, 2020.
Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Clinical information: A 29 year-old woman presented with a 9 cm mass in the thigh without bone involvement. An excision yielded the following specimen.
Gross pathology of the case:
The tumor was lobulated and circumscribed. It had a fleshy gray-white, soft cut surface with scattered small foci that were more translucent than the surrounding tumor tissue.
Histopathology of the case:
|
|
|
|
|
|
| A. | B. | C. | D. | |
|
|
|
|
||
| E. | F. | G. |
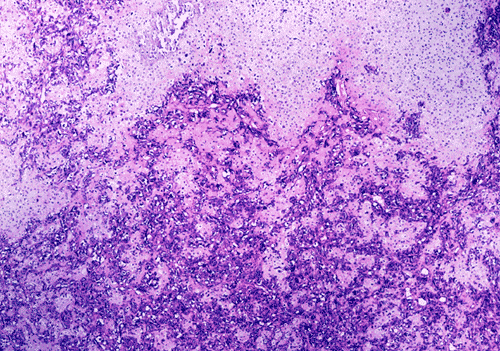
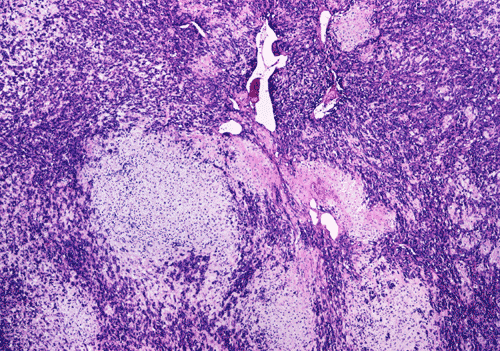
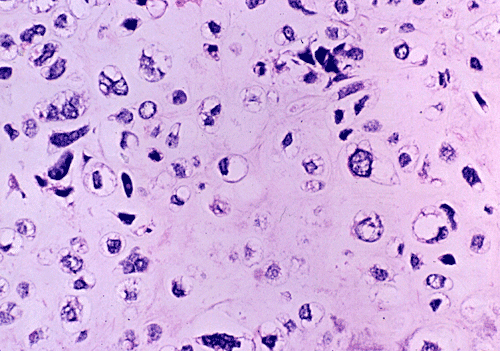
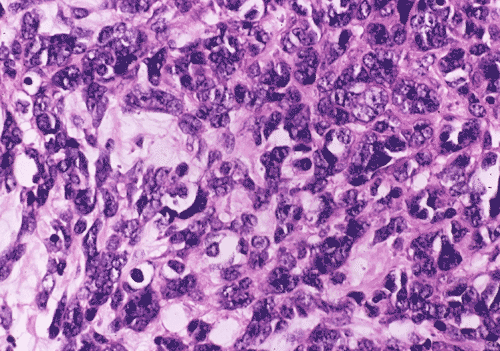
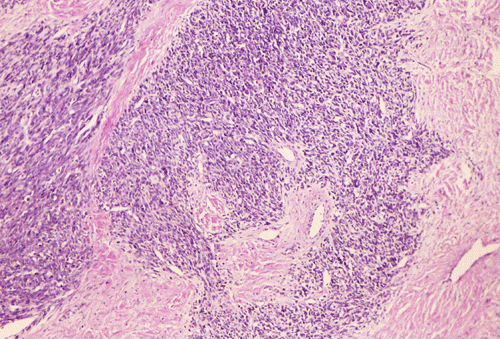
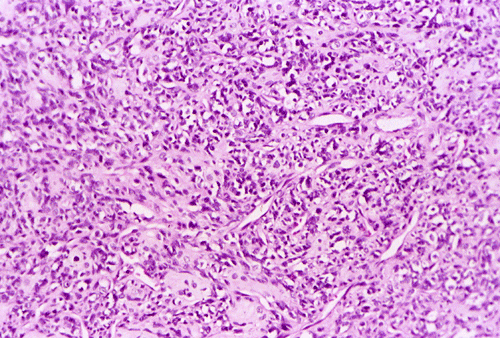
At low power magnification (Panel A and B), the tumor seems to have two distinct components. The first is a highly cellular, basophilic, background stroma. The second is islands of well-demarcated, pale bluish chondroid, hypocellular areas reminiscent of hyaline cartilage. Together, these two components impart a so-called "white clouds in blue sky" histologic appearance. At medium- and high power magnification (Panel C and D), the lacuna in the chondroid areas are well appreciated which helps to confirm the cartilaginous nature of these pale islands. Please note the impressive nuclear pleomorphism in the neoplastic chondrocytes (Panel D).
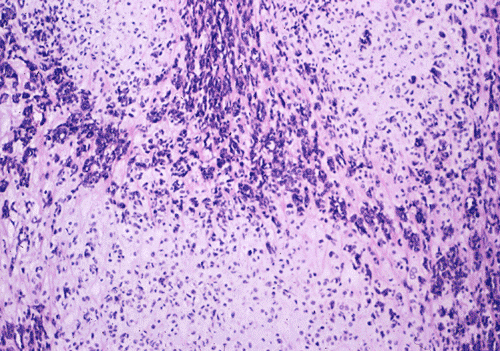
The basophilic background is composed of tightly packed, undifferentiated spindle cells without significant deposition of collagen fibers in between (Panel E). Panels F, G, and H are taken from a different area of the tumor and were not posted on the question web-page. The pathologic change in these areas are very common among mesenchymal chondrosarcoma and should be looked. These include lobules of highly cellular tumor separated by fibrous connective tissue septa (Panel F) and Prominent tumor vascularity with highly branching vessels which (with a bit of imagination!) resemble deer "antlers" or "staghorns"(Panel G and H). This pattern of vessel distribution is similar to that seen in hemangiopericytoma (an uncommon soft tissue tumor) and is therefore often described as a “hemangiopericytoma-like” vascular pattern. The overall gross and light microscopic features in this case are consistent with a diagnosis of primary extraskeletal mesenchymal chondrosarcoma.
| DIAGNOSIS: Extraskeletal mesenchymal chondrosarcoma. |
Discussion: General Information Pathology Molecular Pathology Differential diagnosis
General
Information
Mesenchymal chondrosarcoma is a rare malignant neoplasm that was first
described as a distinct pathologic entity in 1959 by Lichtenstein and Bernstein
1.
Perhaps, it is the most common extraskeletal tumor of osseous origin and it has
a reputation of occurring in odd locations.
Histologically, the tumor is characterized by a bimorphic growth pattern in
which islands of usually well-differentiated hyaline cartilage embedded in a
background of basophilic primitive mesenchymal cells. This created the
well-known “white clouds in blue sky” pictures of these tumors. In addition,
hemangiopericytoma-like components are typically present
[Dabska
M & Huvos AG, 1983]. Mesenchymal
chondrosarcoma differs from conventional chondrosarcomas in three different
aspects. First, it tends to occur in young patients. Second, it has a
predilection for the for the head and neck areas. Third, about one third of
these cases occur in soft tissue. The articles by
Arora & Riddle (2018)
and
Shakked RJ et al. (2012)
are recommended.
Epidemiologically, mesenchymal chondrosarcoma represents less than 2% of all
cartilaginous malignancies [Nakashima
Y et al., 1986]
and typically affects patients in the second and third decades of life, i.e.
adolescents and young adults. However, the tumor has also been reported to occur
in infants and young children particularly in the head an neck region [Dabska
M & Huvos AG, 1983;
Crawford JG et.,
1995;
La Spina M et a., 2003;
Crosswell H et al., 2000]. Males
and females are involved in equal proportion. Mesenchymal chondrosarcoma may
arise in both skeletal and extraskeletal locations, with the former about two
times more common than the latter. In contrast to conventional chondrosarcoma
which typically involve the long bones and pelvic bones, skeletal mesenchymal
chondrosarcomas typically involve the jaws and ribs [Nakashima
Y et al., 1986;
Vencio EF at al., 1998;
Takahashi K et al 1993] whereas
extraskeletal tumors most often occur in the head and neck region, including the
orbit and lacrimal gland [Kashyap
S et al., 2001;
Khouja N et al., 1999;
Kiratli H et al., 2018] and the central nervous system [La
Spina M et a., 2003;
Demirtas E et a;., 2000;
Salvati M et al., 2005], and in the lower extremities, with the thigh being
the most common site. However, this tumor has also been reported in a variety of
unusual sites, including the heart [Nesi
G et al., 2000], femoral vein [Kim
et al., 2003], thyroid [Sadashiva
N et al., 2016] lung
2,
and kidney
3.
Clinical symptoms vary, depending on the site of involvement and whether the
lesion is skeletal or extraskeletal. However, pain and swelling lasting more
than one year is typical. Radiologically, skeletal lesions are primarily lytic
and destructive with poor peripheral margins. Cortical destruction with
breakthrough, and extraosseous extension into soft tissue, are common. Chondroid
type calcifications and foci of low signal intensity with enhancing lobules are
also seen. Most extraskeletal mesenchymal chondrosarcomas are deep seated tumors
with more than two third of them occur in the thigh, particularly the popliteal
fossa
17.
Radiographically, they do not have distinctive features that allows
differentiation from other sarcomas.
Mesenchymal chondrosarcoma is a highly aggressive tumor. In general, they have a
metastatic rate higher than other cartilaginous malignancies and has an
increased tendency to metastasize to the brain, lung, and liver. Metastasis to
the lung is most common. Surgical treatment involves wide or radical resection
of the lesion. Adjuvant radiation therapy has been employed for some high grade
lesions, but systemic chemotherapy has had little or no success. The long-term
prognosis is generally poor with 5- and 10-year survival rates of about 50% and
25%, respectively
2,
3.
Grossly, mesenchymal chondrosarcoma typically appears as a circumscribed,
lobulated, solid mass with a soft, fleshy, grey-white to gray-pink cut surface.
Scattered deposits of cartilage and/or bone of varying size may be grossly
recognized, and areas of hemorrhage and necrosis may be present. Tumor size is
variable with reported tumor diameters ranging from 3 - 37 cm. Microscopically,
mesenchymal chondrosarcomas are biphasic tumors composed of sheets of round to
spindle-shaped primitive mesenchymal cells surrounding discrete islands of
hyaline cartilage. The proportions of primitive and cartilaginous elements vary
widely among tumors and even within different areas of the same tumor.
Cartilaginous differentiation ranges in degree and extent from small foci with
high-grade nuclear features to large areas of well-differentiated cartilage. The
primitive mesenchymal component is highly vascular, typically containing large
numbers of highly branched vascular channels (the so-called
“hemangiopericytoma-like pattern”).
This high variation in the proportion of cartilaginous component and mesenchymal
component must be taken into consideration when core biopsy is employed as a
diagnostic procedure. For a larger tumor, the sample can be dominated by either
component which suggest chondrosarcoma or fibrosarcoma and Ewing sarcoma. With
the high incidence in soft tissue particularly in the head and neck region, a
high index of suspicion is need to avoid misinterpretation of small core
biopsies. On the other hand, this typical white clouds in blue sky pattern makes
diagnosis quite straight forward in resection specimens.
Immunohistochemically, the cartilaginous areas are indistinguishable from other
forms of chondrosarcoma. A wide
range of low- to high-level of differentiation can be seen. Biologically,
different stages of cartilaginous differentiation have been demonstrated in
mesenchymal chondrosarocoma by phenotypic studies [Aigner
T et al., 2000]. The cartilaginous component typically staining
strongly for S-100 protein. However, only isolated cells in the primitive
appearing areas stain for this antigen. NKX3.1 has been shown to be 100%
positive in a small study [Yoshida
KI et al., 2020]. Sox9, a transcription factor considered to be a “master
regulator” of chondrogenesis, was reported to be positive in both the primitive
mesenchymal and the cartilaginous components of 21/22 mesenchymal
chondrosarcomas tested, while being negative in a total of 68 other “small round
blue cell tumors" [Wehrli
BM et al., 2003;
Fanburgh-Smith JC et al., 2010].
The primitive component but not the chondroid component of mesenchymal
chondrosarcoma is immunoreactive for vimentin and CD99 (membranous pattern) [Granter
SR et al., 1996].
In the mesenchymal component, epithelial
markers such as cytokeratin and epithelial membrane antigen are usually
negative. Other than some rare cases [Fanburgh-Smith
JC et al., 2010;
Salvati M et al., 2005],
epithelial
markers such as cytokeratin, epithelial membrane antigen and markers for
rhabdomyosarcomatous differentiation such as desmin, myogenin and myoD are
negative, In over 50% of cases, all components of the tumor i.e. small cells,
lacunar chondroblasts, and chondroid matrix, stain for Leu-7 [Swanson
PE et al., 1990]. INI1 is preserved and this can be used to
distinguish the primitive mesenchymal component from other primitive appearing
tumor with INI1 loss such as atypical teratoid/rhabdoid tumor of the central
nervous system and epithelioid sarcomas [Hollmann
TJ and Hornick JL, 2011]. The small cell component can mimic Ewing sarcoma
and both tumor shows positive membranous staining pattern. FLI-1 is negative in
mesenchymal chondrosarcoma, small cell osteosarcoma, small cell carcinoma, and
rhabdomyosarcoma but positive in 75% of Ewing sarcoma [Lee
AF et al., 2011].
Molecular
Pathology & Cytogenetics:
A recurrent
HEY1-NCOA2
fusion has been demonstrated in 7 out of 11 cases of mesenchymal chondrosarcoma
by FISH [Wang
et al., 2012].
IRF2BP2-CDX1 fusion was demonstrated
in 10 of 15 cases of mesenchymal chondrosarcomas [Nyquist
KB et al., 2012]. These are potential diagnostic markers. A case without
classic histopathological featrues of mesenchymal chondrosarcoma was diagnosed
using FISH for HEY1-NCOA2
fusion [Uneda
A et al, 2020].
Interestingly, there is another case with classic histopathology but lacks both
fusions [Yamagishi
A et al., 2020].
While at least half of chondrosarcoma and chondromas harbors a mutation in
IDH1 and IDH2, mesenchymal
chondrosarcomas do not harbor these mutations [Amary
MF et al., 2011].
On the other hand, mesenchymal chondrosarcoma is positive for NKX3.1 and
AGGRECAN [Yoshida
KI et al., 2020;
Perret R et al., 2020]. This suggest that it is very likely a
NFATc2-sarcomas. This family is characterized by
EWSR1-NFATc2
and fusions and
FUS-NFATc2
fusions [Watson
S et al., 2018;
Wang GY et al., 2019;
Bode-Lesniewska B et al., 2019;
Diaz-Perez JA et al., 2019].
A short list of cytogenetic changes have been summarized [Shakked
RJ et al., 2012].
Translocations involving chromosomes 13 and 21 [der(13;21)(q10;q10)] have been
demonstrated in three tumors (two skeletal, one extraskeletal), possibly
representing a chromosomal rearrangement characteristic of this neoplasm [Naumann
S et al., 2002]. However, a single case of mesenchymal chondrosarcoma has
also been reported to show the reciprocal translocation t(11;22)(q24;q12)
typical of Ewing sarcoma [Sainati
L et al., 1993], suggesting a possible relationship with this entity.
Differential
diagnosis
The histologic differential diagnosis of mesenchymal chondrosarcoma is broad,
and includes dedifferentiated chondrosarcoma,
Ewing's sarcoma, embryonal
rhabdomyosarcoma,
hemangiopericytoma, synovial
sarcoma, small cell osteosarcoma,
and non-Hodgkin lymphoma.
Conventional
chondrosarcoma lacks the primitive mesenchymal component.
Dedifferentiated chondrosarcoma
[Click
here to see a case], however, can
mimic mesenchymal chondrosarcoma. These tumors
typically occur in an older age group and is more likely to affect the
appendicular skeleton. These tumors show an abrupt transition between the
low-grade chondroid component and the high grade dedifferentiated component.
This abrupt transition can suggest mesenchymal chondrosarcoma particularly in
small core biopsies. The dedifferentiated component appears high-grade but not
as primitive mesenchymal component. A hemangiopericytoma-like vascular pattern
is not a feature. Mutations of IDH1
and IDH2 are present in about 50% of
chondrosarcomas, they are not present in mesenchymal chondrosarcomas [Amary
MF et al., 2011].
Ewing's sarcoma
lacks a chondroid component, and tumor cells are typically S-100
protein-negative, unlike mesenchymal chondrosarcoma. Reciprocal translocation
between chromosomes 11 and 22 involving bands q24 and q12, t(11;22)(q24;q12)
leading to
EWSR1-FLI1
fusion occurs in approximately 85% of Ewing’s sarcomas. Other uncommon fusions
include EWSR1-ERG and EWSR1-ETV1 [Source].
Embryonal rhabdomyosarcoma
lacks a chondroid component and consistently expresses muscle differentiation
markers such as desmin, muscle specific actin, and myoglobin. It lacks a
diagnostic gene fusion. Although hemangiopericytoma
and synovial sarcoma may contain areas
with vascular architecture similar to that of mesenchymal chondrosarcoma, they
lack the chondroid component. Small cell
osteosarcoma is typically negative for S-100 protein and have bone
formation. Non-Hodgkin lymphomas are positive
for leucocyte common antigen (LCA) and they also lack the chondroid component.
Reference: