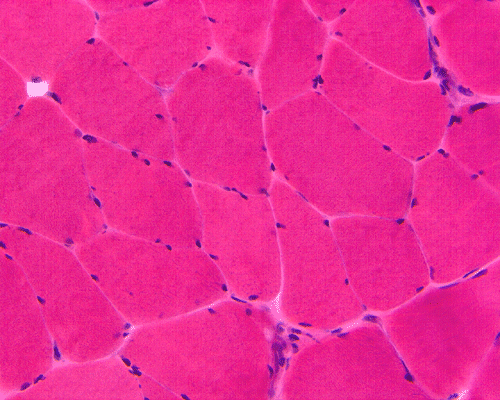


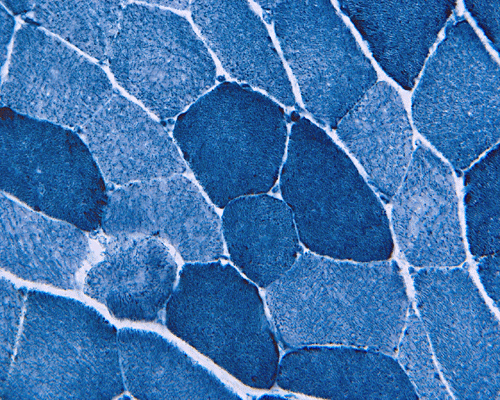


Trichrome
ATPase pH 9.4
NADH-TR
SDH
COX

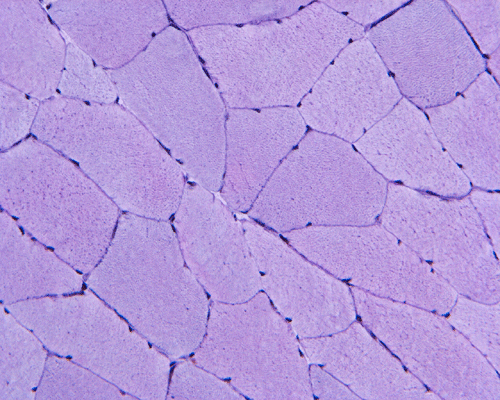
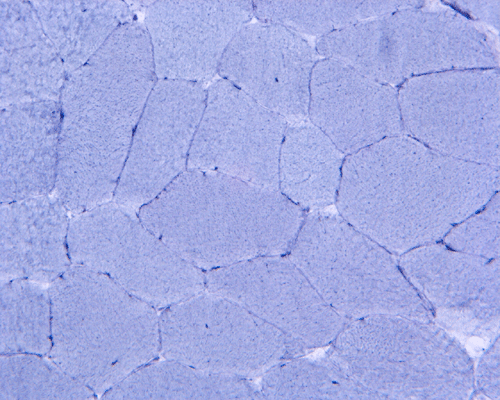

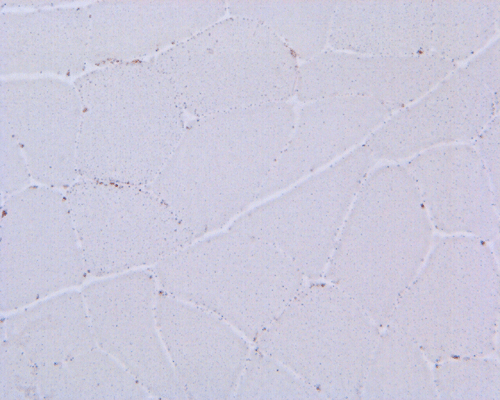
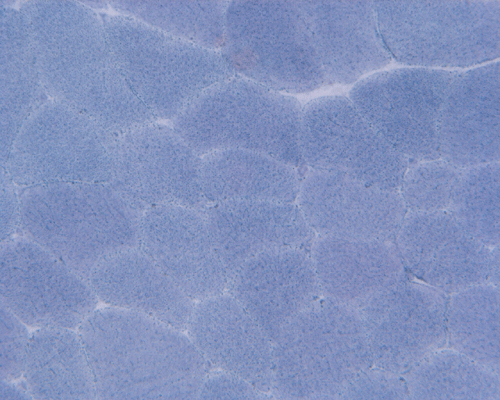
Esterase
PAS
PASD
Phos
AD
AD-Control
| A 33 year-old Man with an
Epidural Mass in the Thoracic Spine. January, 2004, Case 401-1. Home Page |
Syed Shahkhan, M.D.1, Zahid F. Cheema, M.D.1, Kar-Ming Fung, M.D., Ph.D.2 Last update on September 30, 2004
1 Department of Neurology and 2 Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Clinical information:
The patient was a pleasant 56 year-old, right handed white man who presented with chronic insidious weakness of the turncal muscles and proximal muscles of extremities. His problem started about a year ago when he experienced soreness and progressive weakness in the upper arms and thighs. The pain was not associated with exercise or triggered by any known events or activities. He also complained of soreness involving multiple muscle groups along the trunk and torso that extends to the extremities. The patient also described muscle loss in his extremities. His creatine kinase (CPK) in serum was chronically elevated with a highest record of 2380 U/L (normal is 20-140 U/L). There was no numbness or tingling in his hands and feet.
The patient had a history of hyperlipidemia and was treated with antihyperlipidemic drug including Lipitor and Lescol for 10 years but the treatment has been stopped 6 months ago. The patient also had a history of hypothyroidism and was treated by Synthroid but the treatment has been discontinued. At the time of the biopsy, a thyroid function evaluation was within normal limits. There was a history of hypertension and his blood pressure at the time of biopsy was 140/94 mmHg.. There was no history of low exercise tolerance or clauditation. There was no know family history of multiple sclerosis, muscular diseases, or other collagen vascular diseases. The patient was married. He smoked about three cigars per night. He chewed tobacco. He never used any illicit drugs. His father had high cholesterol and coronary artery disease.
On physical examination, his muscle strength and deep tendon reflexes were within normal limits. The rest of the physical examination was within normal limits. Findings on electromyogram (EMG) and nerve conduction studies were within normal limits.
A muscle biopsy was performed as a diagnostic procedure.
|
|
|
|
|
|
|
| A. |
B. Trichrome |
C. ATPase pH 9.4 |
D. NADH-TR |
E. SDH |
F. COX |
|
|
|
|
|
|
|
|
G. Esterase |
H. PAS |
I. PASD |
J. Phos |
K. AD |
L. AD-Control |
| HE | Hematoxylin-eosin stain. | PAS | Periodic acid Schiff reaction. |
| MGT | Modified Gomori's trichrome stain. | PASD | Periodic acid Schiff reaction-diastase. |
| NADH-TR | NADH-tetrazolium reductase reaction. | Phos | Phosphorylase |
| SDH | Succinate dehydrogenase reaction. | AD | Adenylate deaminase |
| COX | Cytochrome C oxidase reaction. | AD Control | Adenylate deaminase control positive |
Pathology of the case:
Hematoxylin and eosin stained sections do not show significant abnormal findings. There was no necrotic fibers or regenerating activity, no inflammatory cell infiltration or evidence of vasculitis (Panel A). The modified Gomori's trichrome stain (Panel B) shows a minute amount of subsarcolemmal deposition which would be normal in this age group. There is no remarkable findings in the ATPase (Panel C), NADH-TR (Panel D), succinate dehydrogenase (Panel E), cytochrome c oxidase (Panel F), and esterase (Panel G). There is some reduction in PAS staining (Panel H). The PAS staining with diastase digestion (Panel I) is within normal limits. The phosphorylase reaction is within normal limits (Panel J). The adenylase deaminase preparation is entirely negative (Panel K) in comparison to the positive controls (Panel L); the reaction is repeated and the same result is obtained.
Comment:
Temporal coincidence suggest that his muscular manifestations is resulted from his antihyperlipidemic medication. However, the current biopsy material does no contain any necrotic or regenerating that could confirm a myopathy second consistent with antilihyperlipidemic drugs. In our experience, patients who are under treatment with antihyperlipidemic drugs and have complaints of muscle, elevated serum creatine kinase and whol went through muscle biopsy do not always have necrotic fibers and regenerating fibers. Therefore, the role of his antihyperlipidemic drug as a trigger of a myopathyic process remain suspicious in this case.
Many patients with myoadenylate deaminase deficiency are asymptomatic and this patient probably falls into this group. The antihyperlipidemic drug, however, may have exert additional stress on his muscle that lead to clinical manifestation. In this particular patient, there is a lack of complaints such as exercise intolerance associated with aches and pain that are more often seen in adenylate deaminase. The significance of adenylate deaminase deficiency as a cause of his clinical manifestation is unclear. The elevated serum creatine kinase, however, is most likely resulted from his antihyperlipidemic drugs.
Isoform determination has not been performed in this particular case. Clinically, however, the deficiency is consistent with myoadenylate deaminase deficiency.
| DIAGNOSIS: Adenylate deaminase deficiency with suspected superimposing HMG co-A inhibitor myopathy. |
Discussion: Myoadenylate deaminase deficiency HMG-CoA reductase inhibitors related myopathy
Myoadenylate deaminase deficiency (MADD): 1
General: MADD is a common enzyme deficiency with a prevalence of 2-3%. The clinical manifestations are heterogeneous and many patients remain asymptomatic.
Biochemistry and genetics: Myoadenylate deaminase (MAD) catalyses the deamination of AMP into IMP and ammonia. IMP can subsequently be re-aminated to AMP. MAD is the muscle-specific isoform of adenylate deaminase. The muscle specific isoform (M isoform) is present exclusively in skeletal muscle. Most of the deficiency is caused by a double transition of cytosine to thymidine at nucleotide 34 and +143 in exon 2 and 3 respectively. The upstream mutation generates a stop codon and lead to the production of a severely truncated protein that cannot be detected by immunofluoresence. The non-muscle specific isoforms (L- and E-isoform encoded by AMPD2 and AMPD3 gene respectively) are present in a variety of tissues including skeletal muscle, smooth muscle, peripheral nerve and endothelial cells. The M-isoform is most abundant in type 2 muscle fiber.
Clinical: The largest group of this deficiency is the asymptomatic and inherited group. Although MADD is asymptomatic in these patients, these patients may develop symptoms when the muscle is subjected to additional stress including medications. MADD may also aggravate the manifestations of a superimposed myopathy such as centrol core disease. The effects of MADD on these patients are not entirely clear.
A smaller group of patients with MADD are asymptomatic. Some of these patients have inherited deficiency. The deficiency of some patients is incomplete but their enzymatic activity is further reduced by associated disorders that lower the enzyme level to deficient range. This is most likely to happen in patients with heterozygous mutations where the level of MAD is reduced but not entirely deficient.
Clinical manifestations are quite diverse but essentially point to metabolic myopathy and include exercise intolerance accompanied by myalgia and cramping. The ratio of blood ammonia to lactate is abnormal after ischemic exercise.
Pathology: MADD does not lead to morphological changes per se. Adenylate deaminase can be easily demonstrated by histochemistry on frozen sections. The reaction is a tetrazolium-based reaction and the reactive product is blue. Biochemical analysis can confirm the deficiency and the isoform. The truncated protein cannot be demonstrated by immunofluoresence.
Anti-hyperlipidemic drug related necrotizing myopathy: 2
General: Necrotizing myopathies can be caused by a variety of drugs including HMG-CoA reductase inhibitors (e.g. lovastatin, pravastatin, simvastatin, and mevastatin), fibric acid derivatives (e.g. clofibrate, gemfibrozil, bezafibrate, and fenfibrate), epsilon aminocaproic acid, etretinate and others.
Clinical: Dyslipidemias can be effectively treated by HMG-CoA reductase inhibitors which specifically inhibit cholesterol synthesis and fibric acid derivatives which are a branched-chain fatty acid esters. A combination of fibric acid derivative and HMG-CoA reductase greatly increase the risk of severe toxic myopathy. Myalgias and elevated serum creatine kinase level may occur.
Pathology: The pathologic changes are that of a necrotizing myopathy with necrotic fibers, regenerating fibers, and some chronic inflammatory cell infiltration. HMG-CoA reductase induced rhabdomyolysis has been described in patients with chronic renal insufficiency, severe hepatobiliary dysfunction, or with concomitant use of gemfibrozil, cyclosporine, erythromycin, or nicotinic acid.
Reference:
Sabina R. Myoadenylate deaminase deficiency in Structural and Molecular Basis of Skeletal Muscle Diseases by Karpati G et al., page 214-215. International Society of Neuropathology, 2002.
Sieb JN. Myopathies due to Drugs, Toxins, and Nutritional Deficiency in Myology by Engel AG and Franzini-Armstrong C, page 1697-1698, McGraw-Hill, 2004.
Cases of the Month Evaluation Coordinator: KarMing-Fung@ouhsc.edu