Squash
Frozen
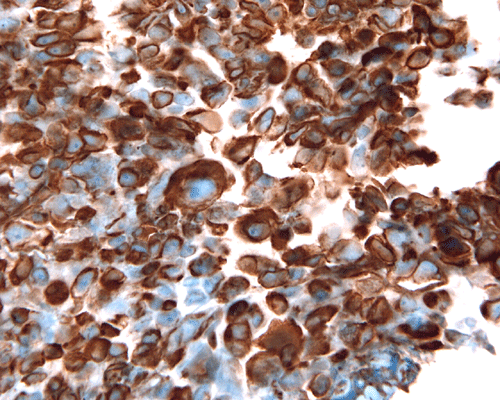
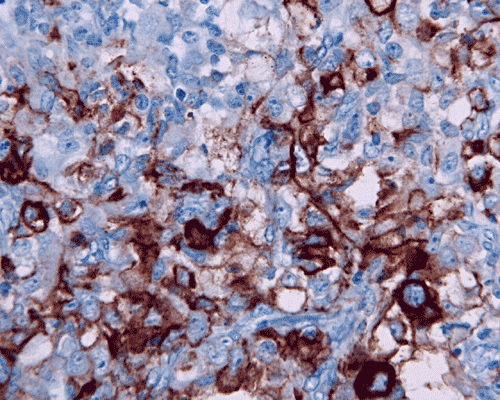
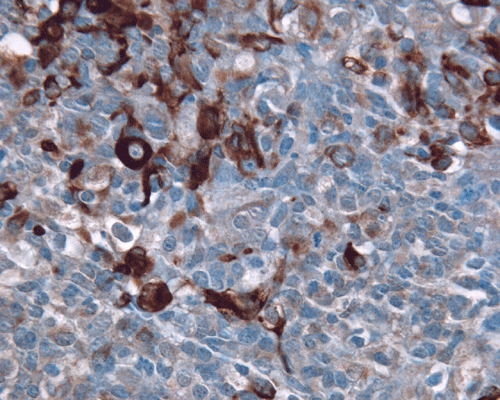
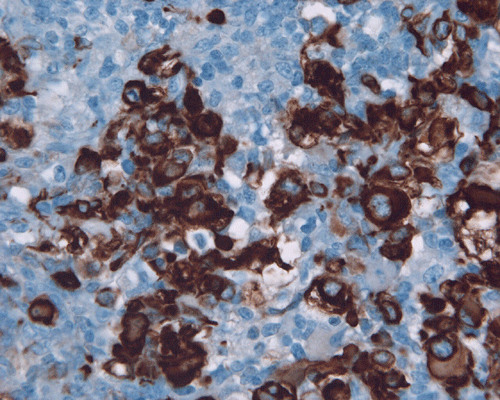
Vimentin
EMA
GFAP
Cytokeratin

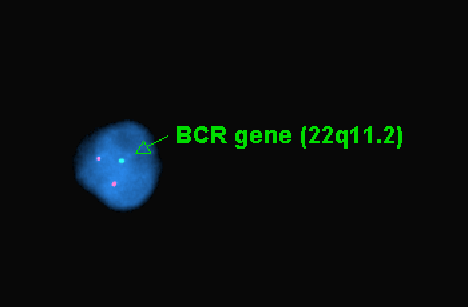
S100
22q deletion FISH
| A Large Cerebellar Mass. March, 2004, Case 403-2. Home Page |
Shibo Li, M.D., Ph.D. 1, Kar-Ming Fung, M.D., Ph.D.2 First posted on May 1, 2004.
1 Department of Pediatrics and 2 Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Kwok Ling Kam, M.B., B.S., FRCPA 1, Kar-Ming Fung, M.D., Ph.D.2 Last updated on on May 25, 2020.
1 Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois
2 Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Clinical information: Click thumbnails to see pictures.
The patient was admitted to our hospital because of a large cerebellar mass that was diagnosed in an outside institution. A surgery was performed. The followings are the representative photos of this mass.
|
|
|
|
|
|
|
|
A. Squash |
B. Frozen |
C. | D. | E. | F. |
|
|
|
|
|
|
|
| G. | H. |
I. Vimentin |
J. EMA |
K. GFAP |
L. Cytokeratin |
|
|
|
||||
|
M. S100 |
N. 22q deletion FISH |
Pathology of the case:
Age of the patient:
|
|
|
|
|
||
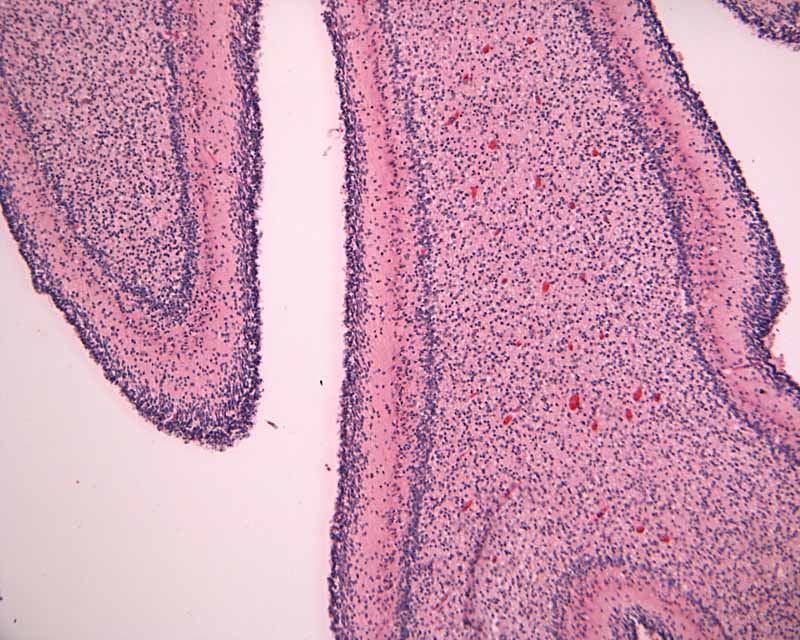
| 1. Lamina dissecans Low-magnification |
2. Lamina dissecans High-magnification |
Intraoperative consultation:
|
|
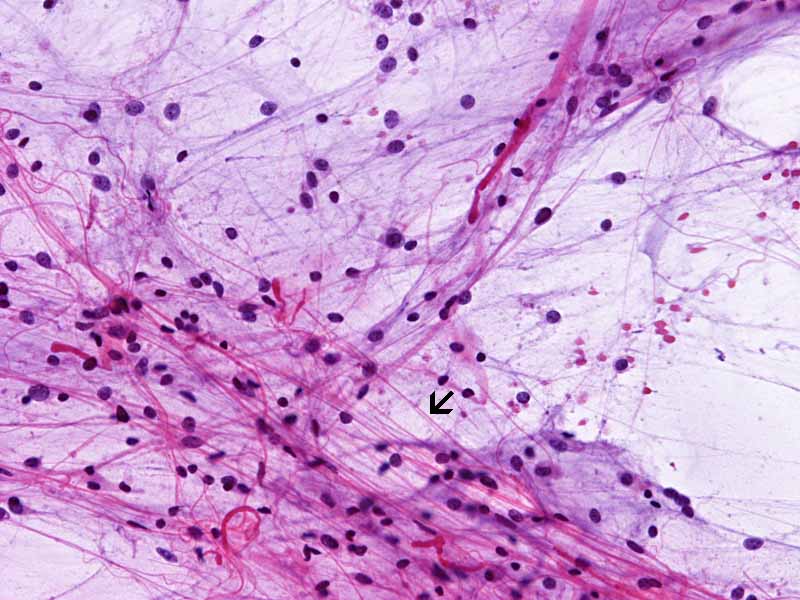
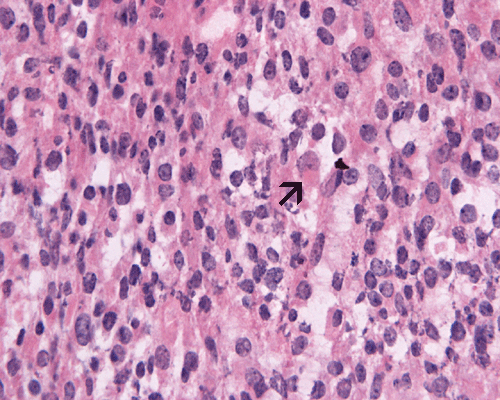
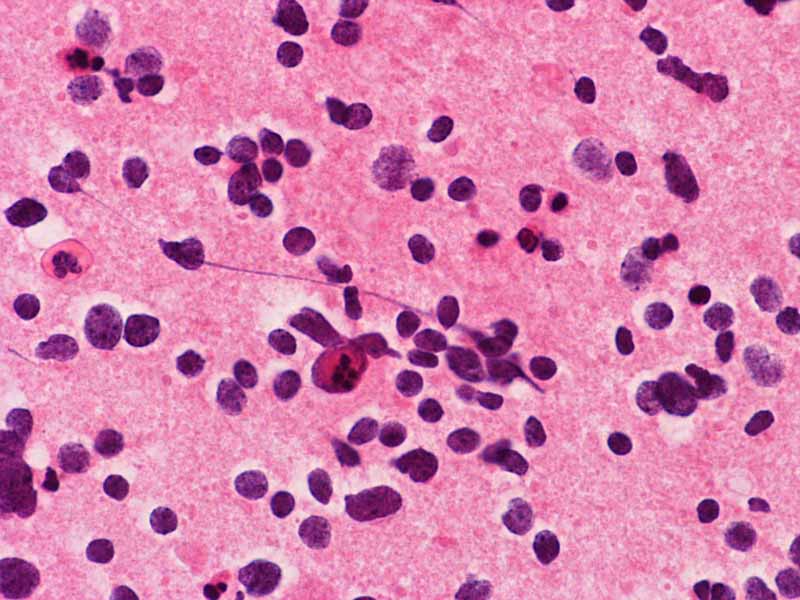
Pilocytic astrocytoma, cytologic preparation. Note the long cytoplasmic processes (arrow). These processes are typically seen in glial neoplasm in particularly the lower grade ones. Note the lack of prominent nucleoli also. |
|
||
|
3. Pilocytic astrocytoma |
||||
 |
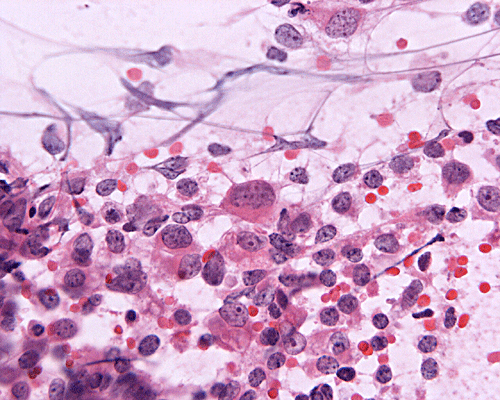
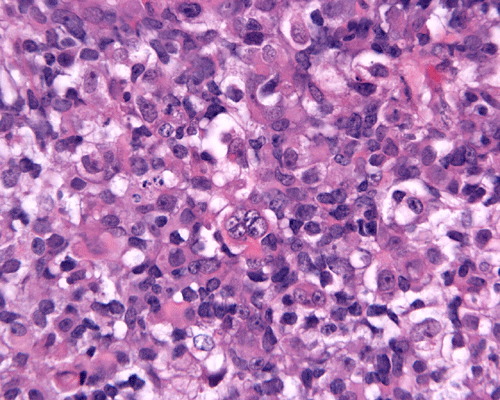
Medulloblastoma, cytologic preparation. Note the lack of prominent nucleoli in comparison to AT/RT in Panel A. One must be alerted that anaplasic/large cell medulloblastomas do contain prominent and may be difficult to be separated from AT/RT with minimal rhabdoid changes. |
|||
|
4. Medulloblastoma |
Permenant sections, immunohistochemistry:
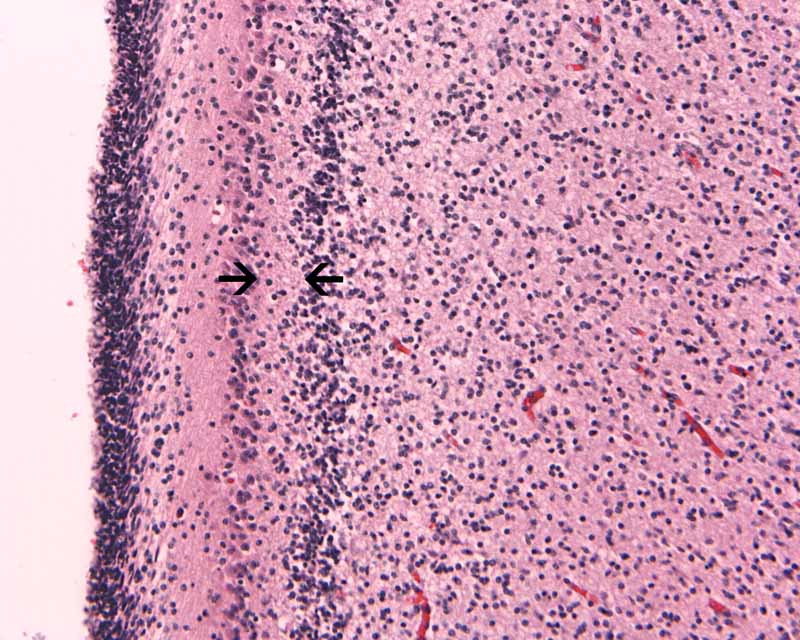
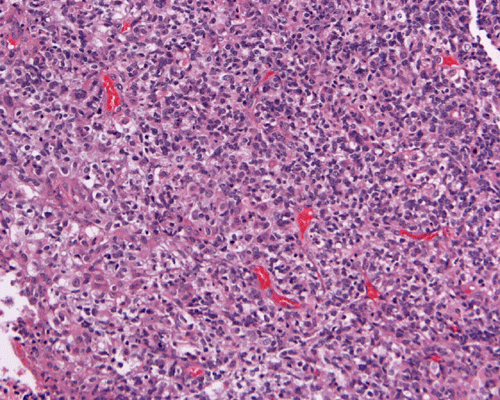
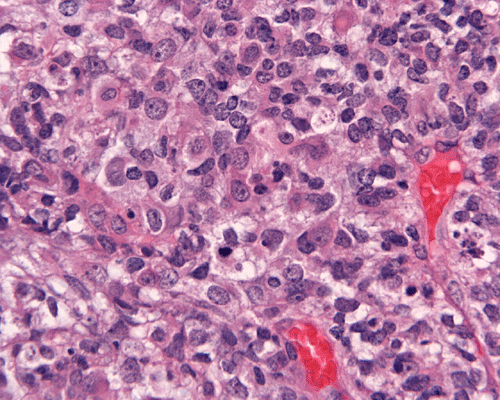
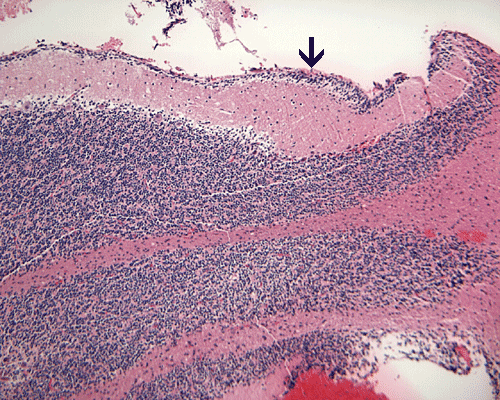
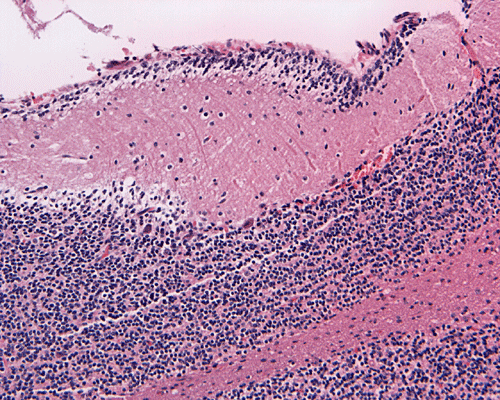
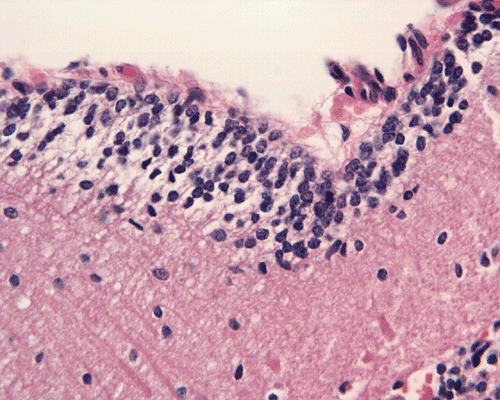
Hematoxylin-eosin stain: The tumor is highly cellular as illustrated in the medium magnification in Panel C. There is a rich vascular supply. Small necrotic foci are present and are not shown here. On higher magnifications as illustrated in Panels D and E, there is substantial nuclear pleomorphism and variation in cell size. Mitotic figures are common. A subpopulation of the cells has substantial amount of eosinophilic cytoplasm and often prominent nucleoli. Some of them have eccentrically located nuclei as demonstrated in the cytologic preparation and some of these cells are multinucleated (arrow in Panel E). Retrospectively, the rhabdoid features are best visualized in the cytologic preparation. These features are highly consistent with an AT/RT.
Cytogenetics:
A piece of fresh tumor tissue was sent for cytogenetics studies at the time of frozen section because of the high suspicion of AT/RT. Routine chromosomal analysis revealed that all twenty cells had apparently normal karyotype. No consistent structural or numerical changes were found. Fluorescence in situ hybridization (FISH) analysis utilizing the BCR gene on 22q11.2 region was also performed, no deletion of 22q11.2 region was detected. However, on interphase FISH of paraffin sections utilizing the same probe as illustrated in Panel N, only one hybridization signal of the BCR gene at chromosome 22q11.2 (green dot) could be detected but the control ABL gene had two dots (red dots). These findings indicated a heterozygous deletion of chromosome 22 involving at least 22q11. 2, which is commonly associated with AT/RT.
Comment:
| DIAGNOSIS: Atypical teratoid rhabdoid tumor (AT/RT), WHO grade IV/IV. |
Discussion: General Pathology Molecular pathology AT/RT and other rhabdoid tumor Differential diagnosis
General Information
Atypical teratoid/rhabdoid tumor (AT/RT) is a malignant primary childhood brain tumor, WHO grade IV as per the 2016 WHO classification of brain tumors. Malignant rhabdoid tumor was first used in 1981 by Haas et al. to describe a highly aggressive childhood renal tumor with cytologic features suggestive of rhabdomyosarcoma [Haas JE et al., 1981]. Bonnin JM et al. described the association of rhabdoid tumors arising in the brain and kidney in children in 1984. According to Weeks et al., about 13% of children with renal rhabdoid tumors developed brain tumors [Weeks DA et al., 1989]. The first rhabdoid tumor of the central nervous system was described by Biggs et al., in 1987. In common to other malignant rhabdoid tumors arising in children and adults, they contain rhabdoid cells as its salient histologic feature.
AT/RT often contains non-rhabdoid components with medulloblastoma-like components and sometime malignant and neoplastic epithelial and mesenchymal components. Rorke et al. published the first comprehensive series of 32 cases on rhabdoid tumors of the central nervous system and also used the term atypical teratoid/rhabdoid tumor (AT/RT) to describe these tumors [Rorke LB et al., 1996] as an attempt to emphasize the diverted histopathologic features in these tumors. Deletion and mutation of the SMARCB1 gene (synonyms: INI1, SMARCB1, hSNF5) in chromosome 22q11.2, and very rarely SMARCA4 which codes the BRG1 protein, are the molecular defining features of AT/RTs [Biegel et al., 2002]. A cell line has been established [Yachnis AT et al., 1998].
AT/RT is an uncommon tumor comprising about 2% or less of all childhood brain tumors and is less frequently seen than medulloblastoma. There is a slight male predominance and most of the tumors occur under the age of two and only rare examples has been reported in adults [Luttenbach J et al., 2001]. Similar to other malignant tumors of the central nervous system, they rarely metastasize outside the central nervous system [Guller E et al., 2001]. The posterior fossa and the cerebellar pontine angle are the most common place of occurrence with a combined incidence of 49% (38% in cerebellum, 11% in cerebellar pontine angle). AT/RT has a distinct tendency to invade into the surrounding tissue in the cerebellar pontine angle. The cerebrum harbors the other 28% of tumors and other uncommon sites including pineal, brainstem, suprasellar, and spinal cord are primary locations of the rest of the tumor (13%) [Rorke LB et al., 1996]. Disseminated tumor is seen in about one third of the cases at presentation. The prognosis is poor [Rorke LB et al., 1996; Burger PC et al., 1998; Bambakidis NC et al., 2002], with a 3-year survival of 22% from the Germany HIT trial center [Biggs et al., 1987]. High dose chemotherapy and radiation can potentially prolong the survival [Meyers SP et al., 2006; Lafay-Cousin L et al., 2012; Geyere JR et al., 2005; Chi et al., 2009]. Rare prolonged survivals have been reported [Hirth A et al., 2003; von Hoff K et al., 2011]. Molecular subgroups of AT/RTs have also been identified using global and transcriptional analysis and the data may help to stratify patients into different risk groups [Torchia J et al., 2006]. MRI findings include variably contrast enhancement, with hyperintensity on FLAIR images and diffusion restriction, the latter is indicative that the tumor is highly cellular [Howlett DC et al., 1987; Meyers SP et al., 2006].
Rhabdoid cells are medium sized to large, oval to round cells. In the most
classic example, the rhabdoid cell contains an eosinophilic, hyaline-like
cytoplasmic globule resembling an inclusion body and occupies a large portion of
the cytoplasm. This hyaline globule displays the nucleus to an eccentric
location. The less classic examples contain a substantial amount of
eosinophilic, often hyaline-like cytoplasm and eccentrically located nuclei.
Although the morphologic features are suggestive of a rhabdomyosarcoma,
cytoplasmic striations typical for rhabdomyoblastic differentiation should not
be present. Marked pleomorphism is seen in most cases and nucleoli are usually
prominent. Multinucleated giant cells are common. Mitotic figures and atypical
mitosis are common. The degree of necrosis is variable but may not be extensive.
Only about 13% of AT/RTs are composed exclusively of rhabdoid cells.
Non-rhabdoid components are seen in the rest. About two third to three quarter
of AT/RTs are associated with medulloblastoma or primitive neuroectodermal tumor
(PNET) like components. Neoplastic epithelial components may occur as
adenocarcinoma-like components, squamous cell with keratinization, or just
simply epithelial cells arranged in nests. Neoplastic mesenchymal components can
also be seen. The amount of rhabdoid cells can vary greatly from uncommon to
substantial. AT/RT with substantial medulloblastoma/PNET like component may be
mistaken as medulloblastoma or PNET [Burger
PC et al., 1996]. Under the electron microscopy, rhabdoid cells
contain bundles of tightly packed intermediate filaments arranged in whorls.
Molecular pathology:
Biallelic inactivation of the SMARCB1 gene (INI1/hSNF5) located in chromosome
22q11.2, either by homozygous deletion or having one mutant allele and the other
allele with deletion or mitotic recombination, accounts for majority of
molecular alterations seen in AT/RT
[Rorke
LB et al., 1996;
SMARCB1
(INI1/hSNF5) is a member of the SWI/SNF chromatin-remodeling complex that
functions in a reversible manner to remodel nucleosomes (i.e. chromatin
remodeling) forming a closed and native state to an open and active state
[
AT/RT and other rhabdoid tumors:
Tumors with malignant rhabdoid phenotype, irrespective of their location and
organ being involved and the age of the patients, behave in a highly aggressive
manner and carry a grave prognosis. Malignant rhabdoid tumors are most
frequently seen in infants and children and are predominantly restricted to the
kidney, the central nervous system (AT/RT) and soft tissue. In adults, malignant
rhabdoid differentiation has been described in many different organs and often
occurs as a component arising in a pre-existing neoplasm including carcinomas
[
Differential diagnosis
Although rhabdoid phenotype has been described in tumors of including meningioma
[
AT/RT with substantial proportion of embryonal tumor component can easily be misdiagnosed [Burger PC et al., 1998]. Rhabdoid cells can usually be found after careful search. One of the key features is the presence of at least distinct if not prominent nucleoli. The rhabdoid change may not be as impressive or dramatic as it appears on text books. A high index of suspicion, immunohistochemical staining with INI1, as well as molecular and cytogenetic studies should be done in suspicious cases, and should be done in all embryonal-looking brain tumors in patients younger than 2 years old. AT/RT merits separation from medulloblastoma and PNETs for its grave prognosis
AT/RT with significant epithelial
component can be mistaken as metastatic
carcinoma. Again, age and clinical history is
very helpful as metastatic carcinoma is extremely rare in infants. The
pathologic features are also different.
AT/RT may show resemblance of
choroid plexus carcinoma.
Both of these tumors occur in young infants and with cerebellar pontine angle a
common site. To make this issue more complicated, deletion of chromosome 22q11.2
has also been described in some malignant childhood tumors that are reported as
choroids plexus carcinoma [Sevenet
N et al., 1999;
Wyatt-Ashmead J et al., 2001]; however, some authors believe that
they may represent intraventricular AT/RT instead. On the other hand, about 40%
of choroid plexus carcinoma shows germline TP53 mutation
Related cases: