T1
T1+Contrast
T1+Contrast
Reticulin
Neurofilament
Vimentin
S100
AE1/AE3
SMA
| A 4 year-old Girl with a
Rapidly Expanding Cervical Intradural, Extramedullary Mass. September, 2004, Case 409-2. Home Page |
Michael Martin, M.D.1, Kalliopi Petropoulou, M.D. 2, Chimène Kesserwan, M.D. 3, Kar-Ming Fung, M.D., Ph.D. 2 Last Update February 28, 2005.
Department of Neurosurgery 1, Department of Radiology 2, and Department of Pathology 3, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Clinical information: The patient was a 4 year-old girl who was in good health. She had difficulties with her left upper extremity for about a week. The problem progressed to her right upper extremity and later to her lower extremities. In a few days, she became quadriplegic. Her sensory functions remained intact. MRI imaging revealed a large intradural, extramedullary contrast enhancing mass that extended from cervical spine C2 to C6 in the anterior aspect of her spinal canal. The tumor also extended through and expanded the foramen on the left nerve roots. Systemic investigation did not reveal any neoplastic disease in other system. The tumor was excised. The followings are representative images of this case.
|
|
|
|
|
|
|
|
|
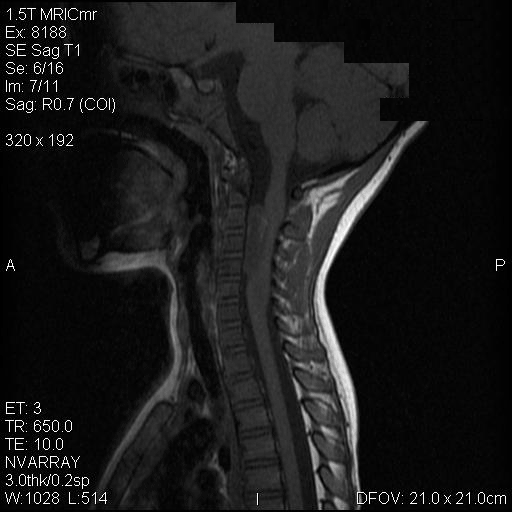
A. T1 |
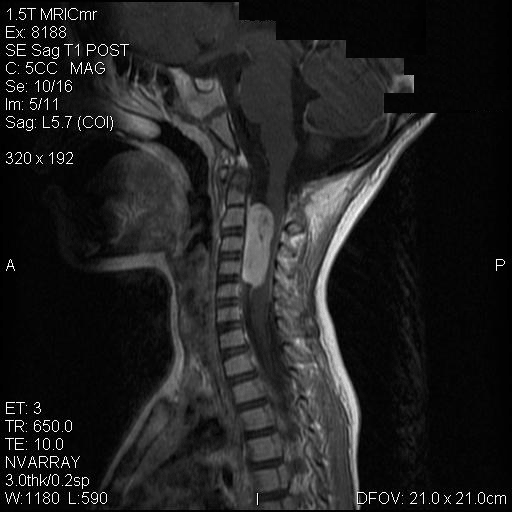
B. T1+Contrast |
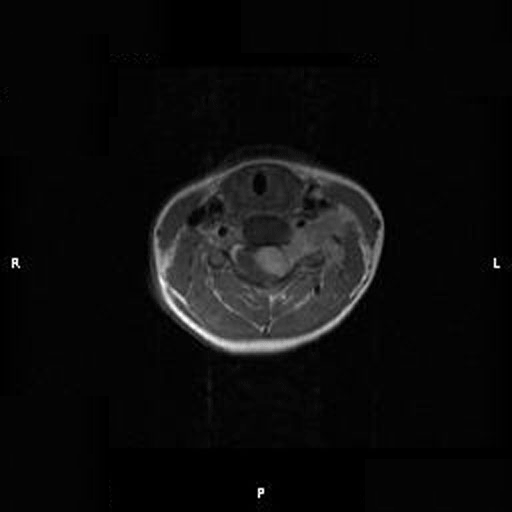
C. T1+Contrast |
D. | E. | F. | G. |
|
|
|
|
|
|
|
|
| H. |
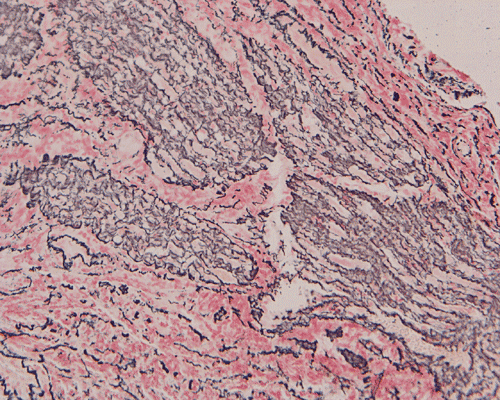
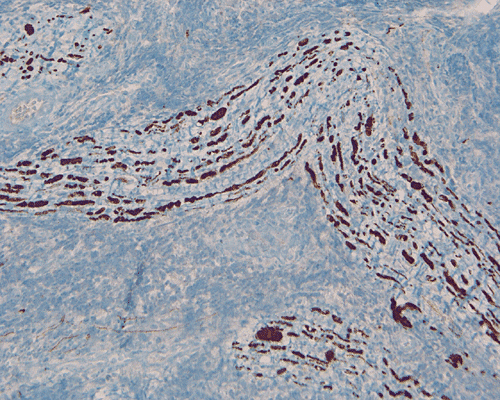
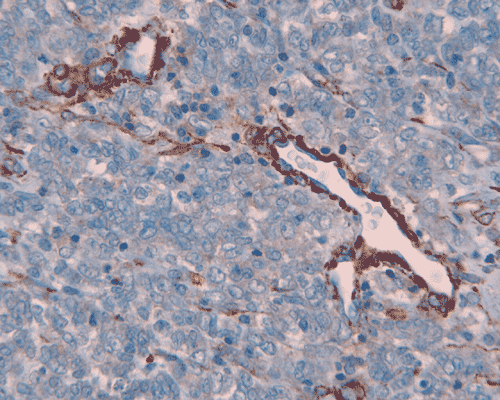
I. Reticulin |
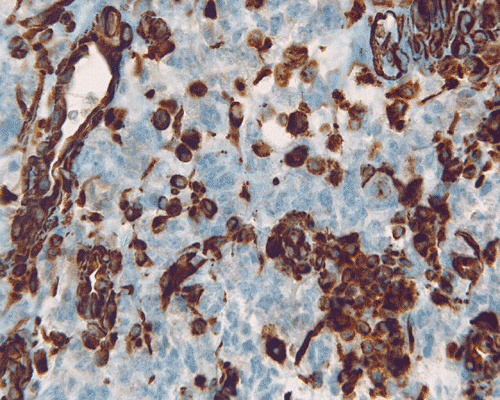
J. Neurofilament |
K. Vimentin |
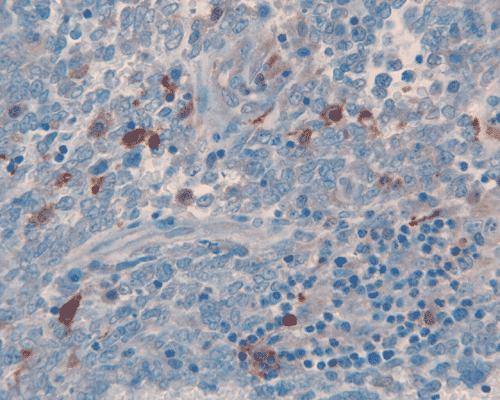
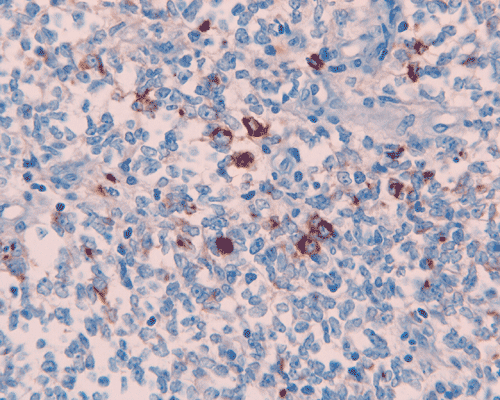
L. S100 |
M. AE1/AE3 |
N. SMA |
SMA: smooth muscle actin
Neuroimaging of the case:
On T1-weighed images, the lesion appeared as a sausage-shaped mass that runs along the long axis of anterior portion of a spinal canal with marked compression of the spinal cord. The mass is well defined from the spinal cord and suggest an intradural, extramedullary location (Panel A). The mass abuts and slightly displaces the left vertebral artery forward. While the intracanalicular portion of the mass is well marginated, the anterolateral, extracanalicular component shows mild infiltration of the musculature. There is slightly heterogeneous enhancement of the mass. Some dural tail enhancement is also present (Panel B). On axial plane, extension of the mass through neuroforamens is well demonstrated (Panel C).
The main differential consideration was nerve sheath tumor. The seemingly rapid progression of the mass is highly suggestive of a high grade tumor. As no lesions are depicted in the brain, the possibility of a drop metastasis of ependymoma, medulloblastoma, supratentorial primitive neuroectodermal tumor (PNET), atypical teratoid rhabdoid tumor (AT/RT), and choroid plexus carcinoma is unlikely. The possibility for a metastatic carcinoma seems remote in this age. There is also a lack of evidence suggesting a metastatic neuroblastoma or hemopoietic neoplasm.
Pathology of the case:
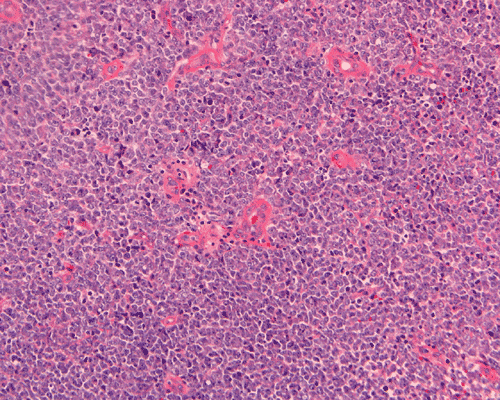
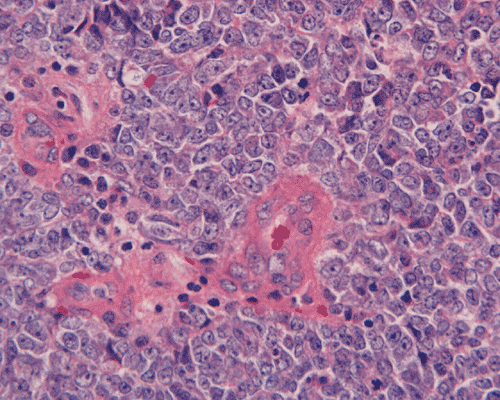
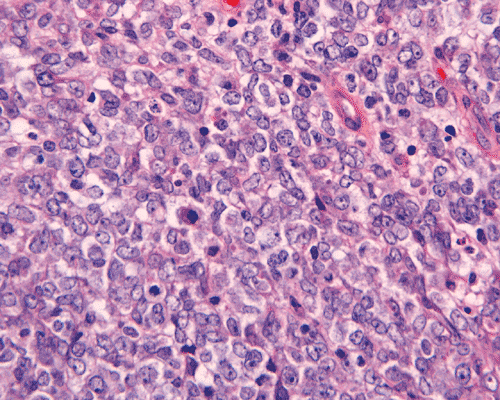
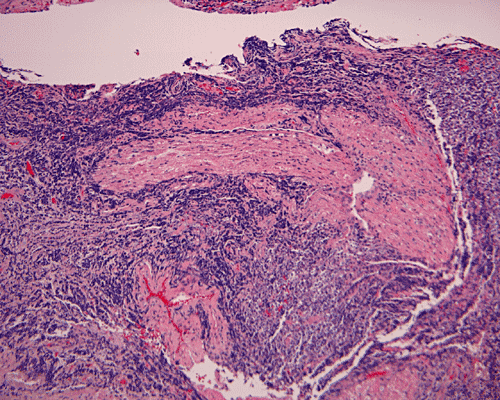
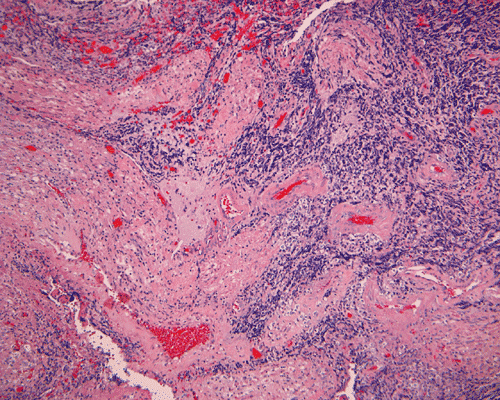
The surgery removed several irregular fragments of tan, soft tissue for examination that were 2.5 x 1.2 x 1.0 cm in toto. Histologically, the lesion is a highly cellular neoplasm with tumor cells packed in solid sheets and without a specific architectural pattern (Panel D). Small, focal necrosis is also present (not shown). The tumor has rich vascularity and the endothelial cells seem to be active (Panel E). Almost all the tumor cells have only a thin rim of amphophilic cytoplasm, large nuclei with prominent nucleoli (Panel E and F). Clear cytoplasm is present in some tumor cells. In general, the cells do not look epithelioid or spindle. In some of the tissue fragment, the tumor shows invasion into the peripheral nerve root with dissection of the nerve fibers (Ú in Panel G and H). This phenomenon is well demonstrated by reticulin stain (Panel I) and immunohistochemistry for neurofilament proteins (Panel J). About half of the tumor cells are strongly positive for vimentin (Panel K). There is also scant positive tumor cells for S100 protein (Panel L) and cytokeratin AE1/3 (Panel M). The tumor cells are negative for neuorfilament proteins (Panel K) and smooth muscle protein (Panel N).
Additional studies:
The first panel of immunohistochemistry that is shown above suggest a high-grade sarcoma with no specific differentiation. The following immunohistochemistry results are further obtained:
|
Antigen |
Result |
Comment |
|
Epithelial membrane antigen (EMA) |
Negative |
Does not support meningothelial or perineurial differentiation. |
|
CD34 |
Negative |
Does not support a diagnosis of solitary fibrous tumor. |
|
CD99 |
Negative |
Does not support a diagnosis of Ewing's sarcoma/peripheral primitive neuroectodermal tumor. |
|
Synaptophysin |
Negative |
Dose not support a diagnosis of tumors with neuroendocrine differentiation including medulloblastoma, primitive neuroectodermal tumor, or neuroblastoma. |
|
HMB45 |
Negative |
Does not support a diagnosis of malignant melanoma. Mostly negative reactivity for S100 protein also does not support a diagnosis of malignant melanoma. |
|
Leukocyte common antigen (LCA), CD20, CD3, CD30 |
Negative |
Does not support a diagnosis of lymphoma. |
|
Myeloperoxidase |
Equivocal |
When interpreted with clinical information and histopathology, the staining result does not support a diagnosis of granulocytic cell sarcoma. |
|
Glial fibrillary acidic protein (GFAP) |
Negative |
Does not support a diagnosis of glial tumor. When the negative result is interpreted with the histopathology, it rules out the possibility of an ependymoma. |
|
Desmin |
Negative |
Does not support a diagnosis of rhabdomyosarcoma. |
Comment:
The tumor under discussion is undifferentiated or poorly differentiated. The scant positive cells for S100 protein cannot strongly support a schwann cell origin. The lack of expression of EMA does not support a peirneurial or meningothelial origin. Therefore, these results do not suppoart a diagnosis of malignant peripheral nerve sheath tumor (MPNST) or meningioma. Both entities are unusual in this age group. While many MPNST may express cytokeratin AE1/AE3, the expression of a scant amount of cytokeratin does not totally rule out the possibility of a MPNST. Although the peripheral nerve bundles are diffusely infiltrated by the tumor as shown in Panel G and H and may suggest a malignant tumor arising from a nerve trunk, there is a lack of solid evidence to prove that this tumor is arising from a nerve trunk. Although a MPNST is suspected, the age and location is quite usual for this entity. The tumor under discussion is best classified as an undifferentiated sarcoma.
| DIAGNOSIS: Undifferentiated sarcoma. |
Discussion: General Information Pathology Diagnostic criteria
General Information
Malignant peripheral nerve sheath tumors (MPNST) refer to a collection of primary malignant tumors of peripheral nerve that are derived from the cells of the perineurium and endoneurium 1. MPNST is an umbrella term that replace the previous term like neuofibrosarcoma or malignant schwannoma. This is mainly because many MPNSTs do not show a full phenotype that allows distinct recognition of them as schwannoma or neurofibroma. MPNSTs comprise about 5% of all malignant soft tissue tumors that arise de novo from normal peripheral nerves or secondary to neurofibromatosis. They are most commonly found in patient between 20 to 50 years old. These tumors are uncommon in childhood and adolescence. MPNST occur only rarely in children younger then 6 years of age. Female are slightly more affected. About 50-60& of MPNSTs occur in patients with neurofibromatosis type 1 (NF1) and the incidence of MPNST in NF1 is 1.9 to 4.6 percent. Plexiform neurofibroma has a particularly high risk for malignant transformation. Larger nerves are more commonly affected than smaller nerves with the sciatic nerve as the most commonly involved. The overall prognosis is poor. Five and ten year survival rates range from 34 to 52 percent, and 23 to 34 percent, respectively, depending upon the series studied 1.
Grossly, MPNSTs are usually covered by a fibrous pseudocapsule, with a gray-tan and firm-appearing cut surface. Some demonstrate bone destruction and approximately 60% show signs of necrosis. In patients with NF1, a pre-existing neurofibroma is often present. MPNST may also demonstrate soft tissue spread, intraneural extension, and osseous invasion.
A variety of histologic pattern may be encountered and thorough sampling is strongly recommended. Microscopically MPNSTs are highly cellular tumor featured by spindle cell proliferation and a high degree of variation, resulting in what is referred to as a “tapestry” appearance. While some tumors show only minor pleomorphism, a moderate to high level of nuclear pleomorphismare often present. Pallisading arrangement of tumor cells as that can be demonstrated in schwannoma is uncommon. Alternating areas of high cellularity and low cellularity can also be encountered. Epithelioid MPNST refer to a group of MPNST that are composed predominantly of epithelioid cells. These tumors tend to arise more often in superficial soft tissue.
Other differentiation with histologic features of malignancy such as rhabdomyoblastic, glandular, squamous, osseous, cartilaginous or neuroendocrine elements are found in about 15% of MPNSTs. Mucin production can be demonstrated in the glandular components of some cases. These tumors must not be mistaken as a metastatic carcinoma with sarcomatoid metaplasia.
Immunohistochemisty for S100 is sensitive but not specific for MPNST. Staining can be patchy or even absent in somce cases. Leu 7 also tends to have a patchy distribution. Although not always present positivity for epithelial membrane antigen (EMA) suggests perineural differentiation. The spindle cells of MPNSTs may express cytokeratin AE1/AE3 but not cytokeratin 7 or 19. In general, MPNST are negative for HMB45 and is an important feature to distinguish these tumors from metastatic malignant melanoma. However, HMB45 is expressed by some MPNSTs and not all malignant melanomas are positive for HMB45. Rhabdomyosarcomatous differentiation can occur and can be well demonstrated by immunohistochemistry for desmin and muscle specific actin.
Electron microscopy is not a helpful diagnostic adjunct in most cases of MPNST.
Diagnostic criteria and this case
As the morphologic and immunohistochemical profile of MPNST overlap with a number of malignant spindle cell tumors, a tumor must meet one of the following criteria 1.
Arise from an identifiable peripheral nerve trunk and have histological features consistent with recognized types of MPNST.
Arise in a patient with known NF1 and show histological features conforming to a recognized type of MPNST.
Arise from an identifiable benign tumor of peripheral nerve, for example pre-existing Schwannoma, neurofibroma or ganglioneuroma.
Show histological features conforming to a recognized type of MPNST, with morphological evidence of Schwann cell or perineurial cell differentiation.
The tumor being present here invaded extensively into the nerve root but a solid proof that this tumor arose from a peripheral nerve root is lacking. On the other hand, there is a lack of definitive schwannian differentiation or features of perineurial cells. Other primary malignant spindle cell tumors of the peripheral nerve have also been described 2,3,4 with some of these tumors associated with radiation therapy 5,6. Although a MPNST is suspected, the tumor under discussion is best classified as a undifferentiated sarcoma.
Reference:
Scheithauer, BW, Woodruff, JM, Erlandson, RA. Atlas of Tumor Pathology, Third Series, Fascicle 24: Tumors of the Peripheral Nervous System. Washington, D.C.: Armed Forces Institute of Pathology; 1999.
Chu PG, Benhattar J, Weiss LM, Meagher-Villemure K. Intraneural synovial sarcoma: two cases. Mod Pathol. 2004 17:258-63.
Dorfmuller G, Wurtz FG, Umschaden HW, Kleinert R, Ambros PF. Intraspinal primitive neuroectodermal tumour: report of two cases and review of the literature. Acta Neurochir (Wien). 1999 141:1169-75.
Mosch A, Kuiters RR, Kazzaz BA. Intradural granulocytic sarcoma: a rare cause of sciatic pain. Clin Neurol Neurosurg. 1991 93:341-4.
Adamson DC, Cummings TJ, Friedman AH. Malignant peripheral nerve sheath tumor of the spine after radiation therapy for Hodgkin's lymphoma. Clin Neuropathol. 2004 23:245-55.
Amin A, Saifuddin A, Flanagan A, Patterson D, Lehovsky J. Radiotherapy-induced malignant peripheral nerve sheath tumor of the cauda equina. Spine. 2004 29:E506-9.
{kind=link}