

MGT
ATPase
NADH-TR
SDH
EST
Oil red O


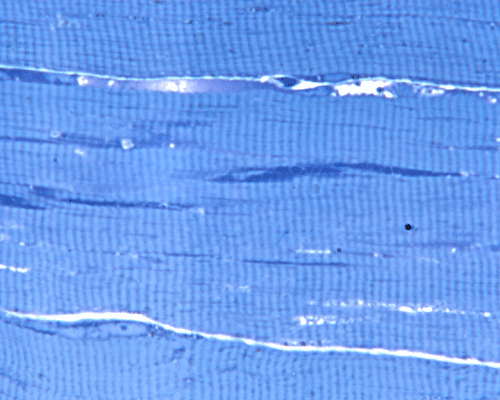

PAS
PASD
AD
PHOS
PHOS Control
Semithin
| A 23 year-old Woman with
Elevated Serum Creatine Kinase Level and Muscle Weakness. November, 2004, Case 411-1. Home Page |
Saud Khan, M.D.1, Zahid F. Cheema, M.D.1, Kar-Ming Fung, M.D., Ph.D.2 Last update on November 30, 2004.
1 Department of Neurology, 2 Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Clinical information: The patient was a 23 year-old woman who was referred for evaluation of muscle weakness and elevated creatine kinase (CK) level in serum. She was a college student. She grew up in a farm and had a tendency to feel tire easily since childhood. She was unable to keep up with her peers in athletic events and heavy farm work. Weakness accompanied by a burning sensation in muscle often occurred shortly after she started to exercise and her symptoms improved with rest. Myalgia would occur after exercise.but there was no experience of tea-colored urine after exercise. Her serum CK level was elevated to two to three times normal on several occasions. On neurologic examination, there was no definitive weakness, muscle wasting, or hypertrophy. An electromyogram (EMG) of her limb muscle was normal. Her thyroid profile was normal. There was no family history of muscle disease. A muscle biopsy was obtained from the vastus lateralis.
|
|
|
|
|
|
|
|
| A. |
B. MGT |
C. ATPase |
D. NADH-TR |
E. SDH |
F. EST |
G. Oil red O |
|
|
|
|
|
|
|
|
|
H. PAS |
I. PASD |
J. AD |
K. PHOS |
L. PHOS Control |
M. Semithin |
N. |
| HE | Hematoxylin-eosin stain. | PAS | Periodic acid Schiff reaction. |
| MGT | Modified Gomori's srichrome stain. | PASD | Periodic acid Schiff reaction-diastase. |
| ATPase | ATPase at pH 9.4 | AD | Adenylate deaminase. |
| NADH-TR | NADH-tetrazolium reductase reaction. | PHOS | Phosphorylase. |
| SDH | Succinate dehydrogenase reaction. | Resin | 1 mm semithin section, touldine blue stain. |
| EST | Esterase | EM | Electron microscopy. |
Pathology of the case:
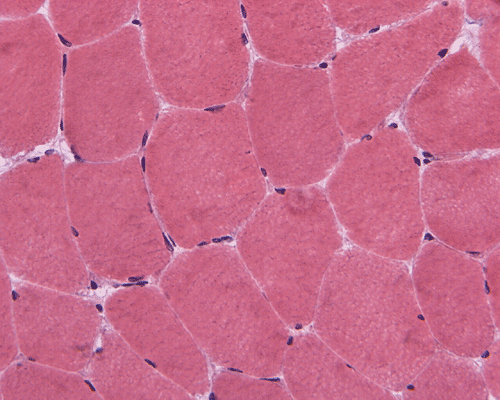
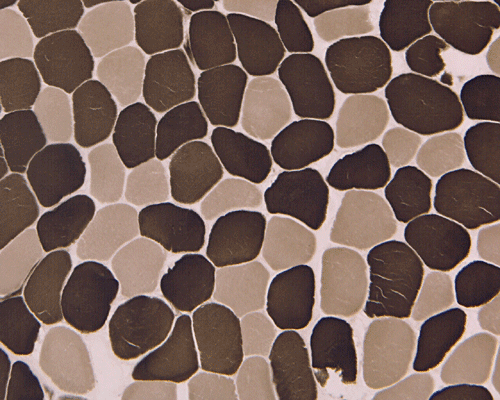
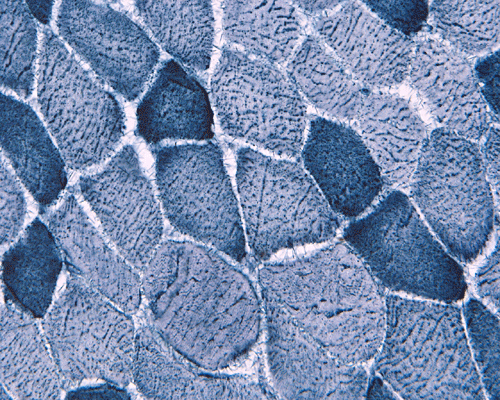
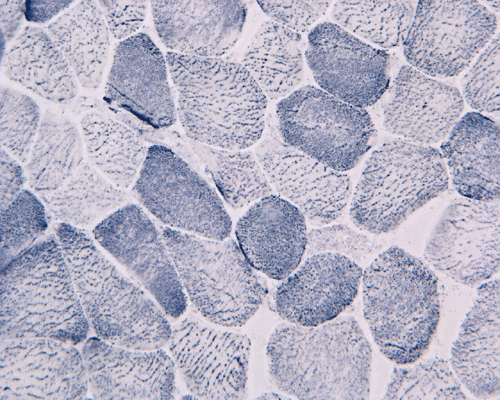
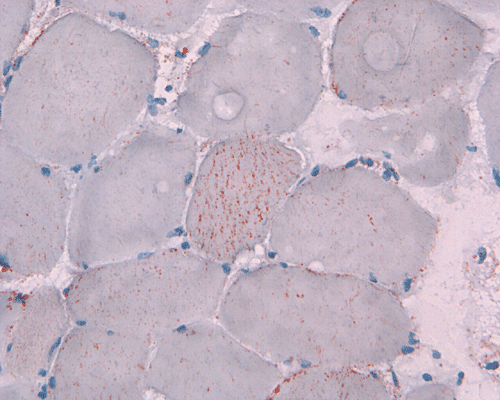
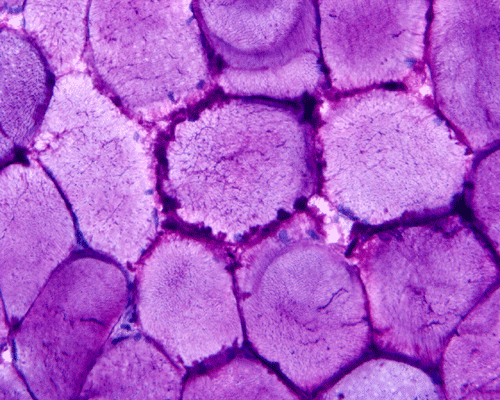
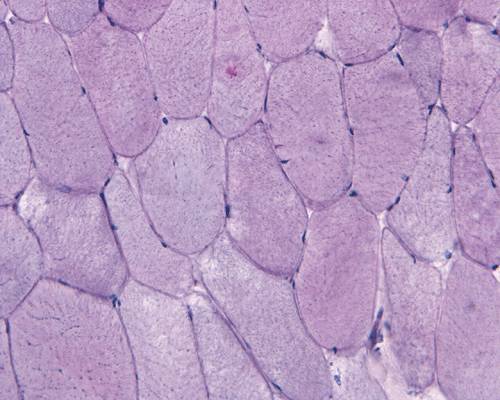
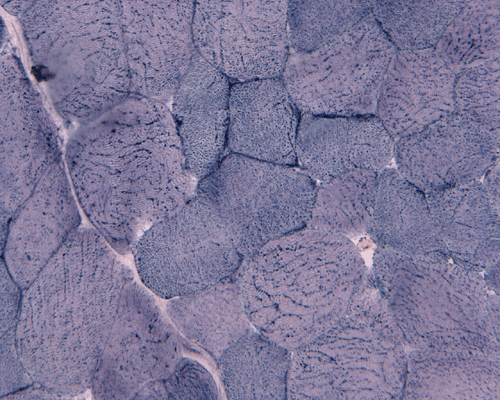
In essence, there are no significant changes demonstrated on the hematoxylin-eosin stain (Panel A) and modified Gomori's trichrome (Panel B). There are no necrotic fibers, regenerating figers, fibrosis, inflammation, increase in centrally located nuclei, subsarcolemmal or sarcoplasmic deposition, or sarcoplasmic vacuolization. On ATPase reaction performed on pH 9.6, type I to type II fiber ratio is in the range of 1:1 and there is no evidence of atrophy or grouping of fibers (Panel C). Type I and Type II fibers can be well distinguished by preparations on oxidative enzymes (NADH-TR and SDH) (Panel D and E). There is no abnormalities on the esterase preparation (Panel F). Occasional fibers with mild increase in lipid content are also present (Panel G). On PAS stain, multiple subsarcolemmal depositions are demonstrated (arrow in Panel H). Most of these depositions can be digested by diastase although a trace of undigested material can be seen (arrow in Panel I). The reaction for adenylate deaminase is within normal limits (Panel J). The reaction for myophosphorylase is almost completely blank (Panel K) when the intensity is compared with the control tissue section (Panel L). On resin embedded semithin sections, trails of dark depositions can be seen (Panel M). On electron microscopy, these materials appear to be coarsely granular material consistent with glycogen (Panel N).
Results of biochemical studies: (Athena Diagnostic, Worcester, MA)
| Enzyme | Reference range (mmol/minute/gram) | Result |
| Glycogen | 0.10-1.50 | 2.27 |
| Phosphorylast A + total | 12.00 | 0.40 |
| Phosphorylase b kinase | 2.43 | 23.83 |
Reference range is 50% of the normal mean.
The level of phosphorylase A is only 3.3% of the normal mean. The amount of glycogen is elevated to 15 times of the high normal. The level of phosphorylase b kinase activity is over 9 times that of normal. These features are typical of McArdle disease.
| DIAGNOSIS: McArdle disease (myophosphorylase deficiency, Glycogenosis type V) |
Comment: Glycogen can be dissolved in aqueous medium during processing and leave subsarcolemmal vacuoles in hematoxylin-eosin stained and modified Gomori's trichrome stained sections. These vacuoles, when seen, should raise a suspicion for glycogenosis (glycogen storage disease). In this particular case, these vacuoles are absent. The diagnosis is first suggested by the negative result in histochemical reaction for myophosphorylase and accumulation of PAS positive, diastase sensitive material. The lack of phosphorylase A activity and increased glycogen content is confirmed on biochemical studies. Phosphorylase b kinase is the enzyme that is responsible to convert the inactive phosphorylase B to the active phosphorylase A. The level of phosphorylase b kinase is typically elevated in McArdle disease.
Discussion: General Glycogenosis Biochemistry Molecular Clinical Pathology
General Information
In 1951, Dr. Brian McArdle described a condition featured by weakness, fatigability, and severe muscle cramping with pain after exercise 1. In 1959, the lack of the myophosphorylase enzyme was identified to be the etiology of this disease 2, 3, 4. McArdle’s disease is a rare disorder caused by deficiency of the muscle isoform of glycogen phosphorylase (myophosphorylase). This is the first myopathy in which a single enzyme defect was demonstrated. The manifestations are entirely restricted to muscle. Other than the fatal infantile form, McArdle’s disease is compatible with long-term survival. It is classically associated with life long exercise intolerance. The initial symptoms are mild and patient may be carrying a label of “lazyness”. Later symptoms include muscle cramps after exercise, myoglobinuria, and, in later course of the disease, muscle wasting. Severe sternous exercise may cause myoglobinuria that is severe enough to cause acute renal failure. Many cases are not diagnosed until adulthood as illustrated in this case.
Glycogenosis (glycogen storage disease):
Glycogenoses are featured biochemically by abnormal metabolism of glycogen. Most but not all subtypes are associated with excessive accumulation of glycogen. Some subtypes lack significant accumulation of glycogen and some are associated with accumulation of abnormal polysaccharide other than glycogen. Thus, glycogenosis is a more appropriate term than glycogen storage disease. There are 11 well known types of glycogenosis as classified by their enzymatic deficiency. Three additional subtypes (Type XII, XIII, and XIV) have only been described in single patient. Many of the glycogenoses have severe and mild clinical forms. Eight of the 11 well known types affect muscle significantly. The liver and the heart are also affected in many subtypes. Type I (Von Gierke disease) and VI (Hers disease) do not affect the muscle. In general, glycogenoses that affect skeletal muscle often share the clinical manifestations of weakness, hypotonia (glycogenosis type II and III), exercise intolerance, fatigue, muscle cramps with or without myoglobinuria after exercise (glycogenosis type V, VII, VIII, IX, X, XI). Glycogenosis type X has significant systemic manifestations but minimal skeletal manifestations.
Glycogen is the primary store of glucose in muscle tissue. Although fatty acid is also used as a major fuel for the muscle, the intracytoplasmic glycogen provides a fast to provide energy and is used before other fuels. McArdle’s disease is caused by a deficiency in muscle isoform of glycogen phosphorylase. This enzyme is involved in converting glycogen into glucose-1-phosphate. Glucose-1-phosphate is in turn converted into glucose-6-phophate to enter glycosis and subsequently into the Kreb cycle and oxidative phosphorylation to generate ATP under aerobic conditions. In McArdle’s disease, excessive chemically normal glycogen piles up as storage material. Since they are normal glycogen, they can be easily digested by diastase on histologic sections. It is important to know that abnormal polysaccharides that cannot be digested by diastase can be found in other glycogenosis (glycogen storage disease). In the absence of adequate supply of fuel, the muscle fibers break down on strenuous exercise. In severe cases, strenuous exercise can result in rhabdomyolysis.
In McArdle’s disease, the glycogen content in muscle fiber is elevated and the activity of phosphorylase is typicall under 10%. Glycogen phosphorylase exist as inactive phosphorylase B and being converted to active phosphorylae A by phosphorylase B kinase. The level of phosphorylase B kinase is typically elevated in McArdle’s diseae.
Molecular pathology:
The gene involved is on chromosome 11q13. The pattern of inheritance is not clear. There are evidence to suggest autosomal recessive inheritance 5 and, less likely, autosomal dominant transmission. The molecular pathology is heterogeneous 6. Mutations lead to the generation of truncated protein and non-functional proteins. Mutation at codon 49 is particularly common in Caucasian patients and account for three fourths of the cases. Molecular diagnosis is possible 7.
Clinical features:
The involvement is completely restricted to skeletal muscle and has a male predominance. Symptoms are highly variable. Patients may be completely normal in between attacks. Although presentation usually begins at childhood and often before 10 years of age, cases that present at elderly age have been described 8. The clinical manifestations are that of metabolic myopathies. The serum creatine kinase level is consistently elevated. The symptoms include fatigue, exercise intolerance, decreased endurance to exercise, exercised induced muscle cramps and pain. Most patients function well once they adjust their activities to a level below their individual threshold for symptoms and learn they can exercise longer if they allow a brief rest immediately after the first sensation of muscle pain. This phenomenon is called the second–wind phenomenon. Strenous exercise can lead to rhabdomyolysis, myoglobinuria and renal failure. Creatine kinase level is the most sensitive marker for rhabdomyolysis. Creatine kinase level peaks within 24-36 hours after initial muscle injury and decreases for 40%/day thereafter.
Generally, three clinical stages are recognized. The early stage occurs in younger patients, namely children and adolescence. In this stage, the patients are easy to get fatigue but some other classic features are lacking. The mid stage is seen in early adulthood and symptoms include muscle cramps, pain, and weakness on exertion accompanied by transient myoglobinuria. In the advanced stage, there is wasting of proximal muscle and weakness. McArdle’s disease is compatible with long term survival. A rare fatal infantile form also exists 9, 10, 11.
The forearm ischemic exercise test (FIET) with serial lactate determinations is used worldwide for the screening of McArdle’s disease and other glucogenosis. Conditions of FIET have been standardized. Taking into account the high sensitivity and specificity of the test, its use should be considered in the screening of anaerobic metabolic myopathies.The test is performed by contracting the forearm to fatigue with a blood pressure cuff inflated to greater than systolic pressure. Antecubital blood samples for lactate and ammonia are collected before and following exercise at 0, 1, 2, 5, and 10 minutes. Ischemia blocks oxidative phosphorylation and ensures dependence on anaerobic glycogenolysis lactate normally rises at least fourfold within 1 to 2 minutes of exercise ammonia rises fivefold within 2 to 3 minutes. The lactate level elevates several-fold under ischemic conditions in normal subjects. There will be no or minimal rise in patients with McArdle’s disease.
The muscle fibers may be of normal sized or even hypertrophic but advanced cases may have small fibers. There is minimal to light glycogen accumulation in form of subsarcolemmal pockets that is PAS positive and diastase sensitive. During processing, the glycogen my dissolve and leave subsarcolemmal vacuoles. This feature, as illustrated in this case, is not always present. Histochemistry will demonstrate a total lack of phosphorylase activity in these otherwise normal looking fibers. Immunohistochemistry for phosphorylase is also negative. Necrotic and regenerating fibers are present in most biopsy specimens. Therefore, it is important not to mistaken these cases as muscular dystrophies. The amount of necrotic fibers may be most abundant if the biopsy is performed several hours after sternous exercise. Most necrotic fibers are likely to be type 2B fibers. Weak phosphorylase activity may be seen in the regenerating fibers. The ultrastructural findings are that of nonspecific necrotic and regenerating fibers. Excessive glycogen with normal structure can be demonstrated. Abnormal mitochondria have been described in some cases. Increased amounts of free and membrane-bound glycogen may be found within axons, Schwann cells, fibroblasts and occasional vascular smooth muscle and endothelial cells that had been included within the skeletal muscle biopsy 12, 13, 14.
Reference:
McArdle, B. Myopathy due to a defect in muscle glycogen breakdown. Clinical Science, 1951 10, 13-33.
Slonim, A. Muscle phosphorylase deficiency (Type V; GSD-V; McArdle’s Disease). Metabolic Myopathies, 65-67.
Schmid, R., Mahler, R. Chronic progressive myopathy with myoglobinuria: demonstration of a glycogenolytic defect in the muscle. Journal of Clinical Investigation, 1959 38, 2044-2058.
Mommaerts, WFHM, Illingworth, B., Pearson, CM., Guillory, RJ., Seradarian, K.. A functional disorder of muscle associated with the absence of phosphorylase. Proc National Academy of Science U S A, 1959 45, 791-797.
Schmidt B, Servidei S, Gabbai AA, Silva AC, de Sousa Bulle de Oliveira A, DiMauro S. McArdle's disease in two generations: autosomal recessive transmission with manifesting heterozygote. Neurology. 1987 37:1558-61.
McConchie SM, Coakley J, Edwards RH, Beynon RJ. Molecular heterogeneity in McArdle's disease. Biochim Biophys Acta. 1990 1096:26-32.
el-Schahawi M, Tsujino S, Shanske S, DiMauro S. Diagnosis of McArdle's disease by molecular genetic analysis of blood. Neurology. 1996 47:579-80.
Wolfe GI, Baker NS, Haller RG, Burns DK, Barohn RJ. Muscle Nerve. 2000 23:641-5.
DiMauro S, Hartlage PL. Fatal infantile form of muscle phosphorylase deficiency. Neurology. 1978 28(:1124-9.
Miranda AF, Nette EG, Hartlage PL, DiMauro S. Phosphorylase isoenzymes in normal and myophosphorylase-deficient human heart. Neurology. 1979 29:1538-41.
Milstein JM, Herron TM, Haas JE. Fatal infantile muscle phosphorylase deficiency. J Child Neurol. 1989 4:186-8.
Byard RW, Lach B, Preston DN. Peripheral nerve and vasculature involvement in myophosphorylase deficiency (McArdle's disease). Pathology. 1991 23:62-5.
Wheeler SD, Brooke MH. Vascular insufficiency in McArdle's disease. Neurology. 1983 33:249-50.
Brumback RA. Ischemia and McArdle's disease. Neurology. 1984 34:559.