| A 2 year-old Girl with a
Funny Looking Eye. November, 2004, Case 411-2. Home Page |
Mason Andrews B.S. (MS-IV)1, Kar-Ming Fung, M.D., Ph.D.2 Last updated November 30, 2004.
1 Fourth year medical student, Class of 2005, College of Medicine, University of Oklahoma and 2 Department of Pathology University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Clinical information: The patient was a 2 year-old girl. She was brought to the doctor because her mother found that her left eye looked "funny". On ophthalmic examination, the pupil of the left eye did not response to light and also has a white reflection. Fundic examination disclosed a retinal tumor. The globe was enucleated. Representative photographs are taken from the surgical specimen.
|
|
|
|
|
||
| A. | B. | C. | D. |
Pathology of the case:
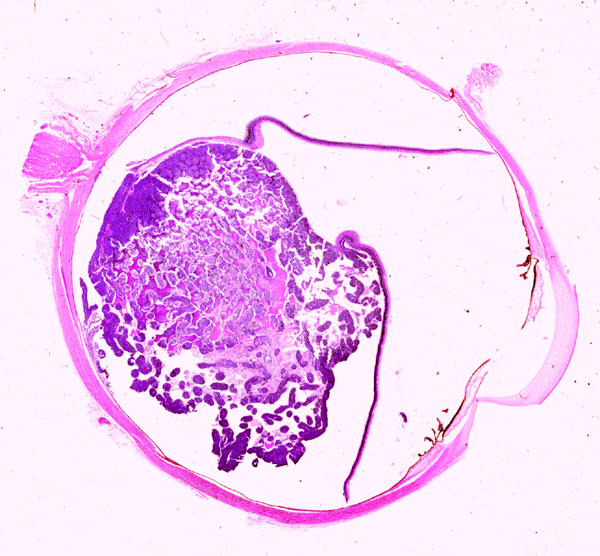
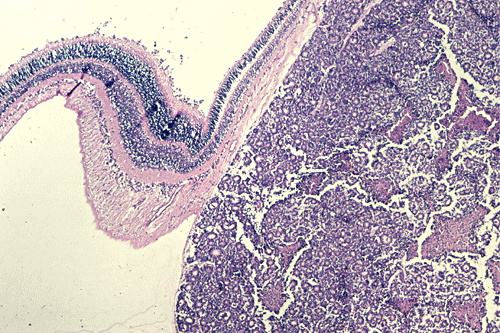
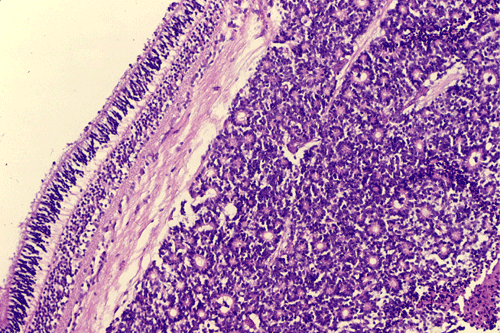
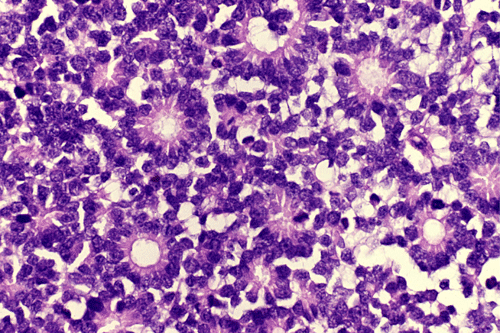
On the whole mount of the globe, there is an exophytic neoplasm that occupies about one-third the volume of the vitreous. The surface of the retina is free of tumor deposition (Panel A). The retina is detached by the mass. There are some pink, necrotic material at the core of the tumor (Panel B). On medium and high-magnification, the tumor is composed of densely packed small cells with hyperchromatic nuclei without prominent nucleoli. A large number of the cells arrange in rossettes with a well-defined luminal border (Flexner-Wintersteiner rossettes) (Panel C and D). The tumor is confined within the globe and the optic nerve is free from invasion.
| DIAGNOSIS: Retinoblastoma. |
Discussion: General Clinical Genetics Gross Pathology Clinical Diff. Pathologic Diff. Experimental Prognosis
General Information
James Wardrop is credited with establishing retinoblastoma as an entity in 1809. He advocated enucleation as a treatment of this disease. Rudolph Virchow later considered the tumor a glioma of the retina and this remained it’s most widely used name for many years. In the late 19th century, Flexner, and Wintersteiner both suggested the tumor be called a neuroepithelioma of the retina. Both described the characteristic rosettes of retinoblastoma which have been given their names. Over the years, many names were proposed, but in 1926 the American Ophthalmological Society adopted the term retinoblastoma which was originally suggested by Verhoeff in 1922 1.
Retinoblastoma is the most common intraocular tumor of childhood 2. The incidence is about 1 in 18,000 live births in the United States. Although relatively uncommon in the general population, it is one of the common childhood malignant tumors. Only uveal malignant melanoma and metastatic carcinoma are more common then retinoblastoma in any age group. Almost 90% of retinoblastomas are diagnosed before 3 years of age with some of them occurring as neonatal or congenital tumors 3. Retinoblastoma occuring after 7 years of age are rare but these tumors can also be seen in adults. There is no particular geopgraphic distribution or sex predilection.
When the tumor is small, it may be asymptomatic and only disclosed on fundoscopic examination. The most common clinical features are strabismus and leukokoria. As most retinoblastomas are seen as hereditary cases, a positive family history in a child with leukokoria should be managed as retinoblastoma until proved otherwise. On CT scan, calcium is often detected. MRI and ultrasound are also useful diagnostic tools particularly in determination of extent of invasion.
Pseudoinflammation or inflammation can be present. Any childhood intraocular inflammation raises the possibility of retinoblastoma and must be worked up accordingly. Orbital inflammation can occur even when the retinoblastoma is confined to the globe and does not indicate extraorbital tumor invasion. Phthisis bulbi, neovascularization of the iris, secondary closed-angle glaucoma, and proptosis can also occur. Leptomeningeal spread and metastases can occur in the later course of the disease.
If left untreated, retinoblastoma will fill the eye and completely destroy the internal architecture of the globe. It will invade through the optic disc into into the optic nerve and further disseminate. Extraocular extension is the rule in advanced, untreated case that are far more commonly seen in the past. The tumor may present as a fungating mass prolasping through a corneal perforation. Proptosis secondary to massive posterior orbital invasion, buphthalmus, neurological manifestations related to brain or leptomeningeal involvement, and enlarged preauricular or submandibular nodes would be present. These clinical presentation, however, is becoming uncommon as early detection is achieved in many cases.
Genetics and Molecular Pathology
Based on statistical analysis, Knudson postulated the two-hit hypothesis stating that retinoblastoma requires two tumor inducing events to convert a normal retinal cell into a neoplastic cell. In cases of heritable retinoblastoma, the first hit is in the germ cell and the second occurring later after birth. In non-heritable retinoblastomas, both hits occur after birth 4, 5. He estimated the number of tumors per patient with a heritable mutation to be between 3-4. Also of notable significance is that survivors of the heritable type are highly susceptible to the development of other non-ocular cancers. The retinoblastoma gene (RB1) is identified to be located at chromosome 13q14 and is the first human cancer for which a susceptibility gene was isolated and sequenced 6. Current evidence indicates that the RB1 gene is a tumor suppressor gene. Although RB1 is involved in malignant progression in many different tumors, its role as an initiation is restricted to the retina. The retinoblastoma gene deletion syndrome is inherited as an autosomal dominant trait and is featured by retinoblastoma in the retina, often multifocal and bilateral, with or without an intracranial tumor that is most often seen in the pineal area (trilateral retinoblastoma), increased incidence of second malignancy other than retinoblastoma, multiple congenital abnormalities and mental retardation.
Special Features of Retinoblastomas
Several aspects are unique to retinoblastoma. First, these tumors can be divided into hereditary and non-hereditary (sporadic) type. In the hereditary type, about 25-30% of the retinoblastomas are bilateral. Multifocal tumor within the same globe is also a strong indication for hereditary type.
Spontaneous regression of retinoblastoma has been well documented 7, 8, 9. The incidence of spontaneous regression is estimated to be about 2% 10. Although the regression can be bilateral 10, it is more often unilateral and has been reported in identical twins 11.
Trilateral retinoblastoma refers to the association of a midline intracranial neoplasm with bilateral retinoblastoma. This association was first recognized by Jakobiec and Tso in 1977 12. Trilateral retinoblastoma is very rare and is observed only in hereditary retinoblastoma. The tumor is often a primitive neuroectodermal tumor arising in th pineal gland. Patients often die of cranial spinal dissemination of the primitive neuroectodermal tumor despite that it produces minimal mass effects 13, 14. It is also important to notice that trilateral retinoblastoma can manifest as unilateral retinoblastoma and intracranial tumor 14, 15, 16. A suprasellar tumor rather then a pineal tumor is more likely to be seen in trilalateral retinoblastoma associated with unilateral retinoblastoma 14.
Second malignant neoplasm, usually a sarcoma, is common in survivor of hereditary type of retinoblastoma 17, 18 and the estimated frequency is about 15% to 20% 19.
Gross Pathology
At the early stage, the tumor appears as a white nodule or multiple nodules (multicentric tumor) in the retina. The small nodules may suggest military tuberculosis involving the retina on fundoscopic examination. In bilateral cases, tumors often present simultaneously or with only a short time lag between tumors arising from the two globes.
As the tumor grow into a mass, some of them have an endophytic pattern that grows from the inner surface of retina into vitreous with tumor cells shedding into the vitreous. The tumor cells will disseminate and coat the retinal surface. As retinoblastoma in hereditary cases can be multicentric, such retinal coating should be distinguished from multifocal retinoblastoma. Retinal seeding is suggested if the tumor lies mainly on the surface or if clusters are seen within the vitreous. Tumor cells from the vitreous may also reach the anterior chamber.
The exophytic type grows primarily from the outer retinal surface of the retina towards the choroids and is well illustrated in this case. The tumor will produce an elevation and then a detachment of the retina. From the choroids, tumor cells invade into the orbit and conjunctiva. Extraocular extension and metastasis will follow. Neither of these patterns carry a prognostic significance. A mixed pattern that consists of both the endophytic type and exophytic type is perhaps the most common type.
The diffuse infiltrating type 20, 21, 22 is the least common and most difficult to diagnose clinically. These tumors infiltrate the retina diffusely without thickening it. Cells can also shed into the vitreous to seed the anterior chamber and produce a pseudohypopyon. When this happens it may masquerade as retinitis, uveitis, vitritis or Toxocara endopthalmitis on clinical examination. This type also has several features that distinguish it from other retinoblastomas. Diffuse infiltrating retinoblastomas are rare and are unilateral in all the reported cases, occur in older children with an average age of 6 years, more common in boys, no case of second tumor have been described 20, and mostly sporadic with only one hereditary case reported 23,
In the rare event of spontaneous regression, the tumor is reduced to a necrotic and/or gliotic and partially calcified residue at the site of the neoplasm.
Microscopic Pathology
The pathology is typical for that of a small blue cell tumor with morphologic and immunohistochemical features comparable to medulloblastoma, supratentorial primitive neuroectodermal tumors, and adrenal neuroblastomas. Tumor cells have small to medium sized, hyperchromatic nuclei with clumpy chromatin and without prominent nucleoli. Only a small to minute amount of cytoplasm is present. Mitotic figures are numerous. Retinoblastoma has a striking tendency to undergo necrosis. The tumor cells that are closer to the blood vessels are more likely to remain viable. Calcifications are common findings in the necrosis. Some of the nucleic acid liberated from these necrotic cells becomes absorbed in the walls of blood vessels giving them a deep blue appearance.
Most often, tumor cells arrange in Flexner-Wintersteiner rosettes typified by tall cuboidal cells formaing a small, circumstribed lumen with well defined limiting membrane on the luminal side. The tumor nuclei are found at the basal end of the cuboidal cells while the apical end contains the cytoplasm. Small projections are protruding from the luminal surface into the lumen and these projections represent residual features of photoreceptor cells. Homer-Wright rosettes can also be found but are less common. In between the rosettes are solid sheets of poorly differentiated tumor cells.
Fleurettes are small clusters of tumor cells featured by long, eosinophilic, cytoplasmic processes, and bland cytologic features. They may arrange like a bundle of flowers and therefore its name. The fleurettes contain structures that resemble retinal cone cells and indicate differentiation along the photoreceptor lineage. Fleurettes may be absent, present in small nodules, or occasionally comprising a significant volume of the tumor. Cells forming the Flexner-Wintersteiner rosettes and fleurettes are considered more differentiated than remaining tumor cells.
In the occasional case of complete regression, the tumor is turned into a gliotic mass with necrosis, calcification but without viable tumor cells.
Immunohistochemistry, the tumor cells are positive for antigens that are expressed by neuronal cells such as synaptophysin and neurofilament proteins 24 as well as antigens that are expressed by photoreceptor cells such as S-antigen and rodopsin 25, 26, 27 are detected in retinoblastoma cells to a variable extent. Expression of S100 protein and glial fibrillary acidic protein (GFAP) are also demonstrated and is regarded as expression by reactive cells by some investigators 24 and expression by tumor cells by other investigators 27. At the ultrastructural level, features suggestive of differentiation into a photoreceptor including cilia with a 9+0 pattern of microtubule doublets typical of photosensory neurons, large numbers of cytoplasmic microtubles, synaptic ribbons, and dense core (neurosecretory) granules are present.
There is a family of small, often calcified tumors that are typically found in eyes retaining useful vision and have fundoscopic appearance similar to retinoblastomas that have been treated by radiation. Some investigators consider these tumors to be spontaneously regressed retinoblastoma 7 and other investigators consider them the benign version of retinoblastoma and called them retinocytoma or retinoma 28.
Clinical Differential Diagnosis
The list of clinical conditions that can simulate retinoblastomas is long. The three common entities that can be easily mistaken as retinoblastoma are Toxocara endophthalmitis, persistent hyperplastic primary vitreous and Coat’s disease. Pathologically, these entities should not be confused with retinoblastomas.
Toxocara canis endophthalmitis is a parasitic disease that clinically mimics endophytic retinoblastomas. It is almost always a disease of children but is never present at birth. The ocular lesions are painless, often without external signs or symptoms. The larvae do not invoke an inflammatory response until they die and then a characteristic eosinophilic abscess forms around the degenerating organism. The most frequent histologic finding is a sclerosing vitreitis that has caused a total retinal detachment. Plasma cells are the most common cells in the infiltrate. Eosinophils may be rare unless the section is close to a degenerating Toxocara larva.
Persistent hyperplastic primary vitreous is a congenital anomaly of the primary vitreous that often mimics exophytic retinoblastoma clinically. The vascular components of the primary vitreous normally regress during the third trimester disappears on birth. In persistent hyperplastic primary vitreous, these vessels persist after birth and are associated with proliferation of mesenchymal tissue. Most often, this tissue is located behind the lens and is firmly adhered to the posterior lens capsule and is often associated with retinal detachment and retinal dysplasia.
Coats' disease is characterized by a peripheral vascular telangiectasis of the retina. These vessels leak large amounts of exudates that accumulate in the outer retinal layers and subretinal space leading to a total retinal detachment. The exudate is PAS positive and rich in lipid. Foamy macrophages and cholesterol clefts frequently accumulate in the subretinal exudates. In contrast to Toxocara endophthalmitis and persistent hyperplastic primary vitreous, there is neither sclerotic change nor mesenchymal proliferation in the vitreous.
Pathologic Differential Diagnosis:
Medulloepitheliomas, also know as “diktyomas” in the past, are rare retinal tumors that display morphologic and phenotypic features recapitulating the sixth week stage of development of the optic vesicles. In contrast to medulloepitheliomas arising in the brain that carry a grave prognosis, there were only 4 fatal cases out of 61 cases that have been reported in two separate series 29, 30. These rare tumors occur in later childhood with a mean age of 5 years at the time of definitive diagnosis 29, 30. Histologically, there is a benign form and a malignant form. These tumors usually arise from the ciliary epithelium as a well circumscribed mass or it may be infiltrating the surrounding tissue. Rarely, they are found in the iris or optic disc. On low-magnification, these tumor may have a lacy or net-like architecture.Most often, they have an organoid pattern of growth and the epithelioid cells have a limiting membrane in the inner, free surface. Mitotic figures are common. These epithelial cells are supported by a stroma. The teratoid variant (teratoid medulloepithelioma) contains heterogenous components such as cartilaginous, osseous, and rhabdomyoblastic components. The malignant form contains undifferentiated round cell component. Solid areas of these tumors resemble retinoblastomas. The other criterion for a diagnosis of malignancy is overt sarcomatous changes in the heterologous components.
Retinal dysplasia is often but not always associated with trisomy 13 (Patau’s) syndrome which are congenital and bilateral. Unilateral cases are not associated with trisomy 13. Histologically, there are tubular and rosette-like structures in the dysplastic neural retina. In those associated with trisomy 13, cartilage can be found in microphthalmic eyes smaller than 10 mm. The rosettes can have three layers, two layers, or one layer of moderately to well differentiated neural cells. Primitive unilayer rosettes formed by a single layer of undifferentiated neural retinal cells surrounding a lumen with a tangle of fibrils can also occur. There is no tumor formation in retinal dysplasia.
Astrocytomas of the retina are rare. They are often associated with tuberous sclerosis and less frequently with neurofibromatosis 31. Longstanding lesions may be calcified that would clinically and radiologically suggestive of retinoblastoma. Histologically, these tumors have features of astrocytomas and the cells strongly express GFAP. They should be easily distinguished from retinoblastoma. Glioma arising from the optic nerve or optic nerve head may have intraocular protrusion. Many of these cases are associated with neurofibromatosis 1.
Malignant melanoma arising from the uvea occurs in middle aged adults with mean age of 55 years. They are far more common in white than black patients. Bilateral or multifocal tumors are extremely. The tumor cells can be spindle, epithelioid or a mixture of both patterns. Demonstration of melanin, when it is present, is a helpful clue. Immunohistochemistry, melanoma are strongly and diffusely positive for S100 and often positive for HMB45.
Experimental Studies
Transgenic mouse models of retinoblastoma generated by Simian virus 40 (SV40) large-T antigen as the oncogene are available 32, 33, 34. Interestingly, SV40 large-T antigen has also generated degeneration of photoreceptor cells 35 and Purkinje cells in cerebellum 36 without tumor production in both models. In another murine model using SV40 large-T antigen, degeneration of cells expressing SV40 large-T antigen precedes tumor formation 37. The interesting relationship between cell proliferation and cell death and tumorigenesis in SV40 large-T angiten carrying transgenic mouse model has been reviewed by Fung and Trojanowski 38.
Recurrence after enucleation of the globe is almost always secondary to tumor cells left in the orbit or optic nerve. With proper treatment, the 5-year cumulative survival rate is 91% in the United States 39. Second malignant neoplasm and trilateral retinoblastoma become important burden in hereditary retinoblastoma 17, 18. When metastases occur, they typically occur within the first 1-2 years after treatment. Death caused by metastases after this period is quite uncommon.
Reference:
McLean IW, Burnier MN, ZimmermanLE, JakobiecFA, Tumors of the Eye and Ocular Adnexa, Armed Forces Institute of Pathology, Washington D.C., 1994, pp. 97-154.
Castillo BV Jr, Kaufman L. Pediatric tumors of the eye and orbit. Pediatr Clin North Am. 2003 50:149-72.
Moore SW, Satge D, Sasco AJ, Zimmermann A, Plaschkes J. The epidemiology of neonatal tumours. Report of an international working group. Pediatr Surg Int. 2003 19:509-19.
Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971 68:820-3.
Knudson AG Jr, Meadows AT, Nichols WW, Hill R. Chromosomal deletion and retinoblastoma. N Engl J Med. 1976 295:1120-3.
Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987 235:1394-9.
Aaby AA, Price RL, Zakov ZN. Spontaneously regressing retinoblastomas, retinoma, or retinoblastoma group 0. Am J Ophthalmol. 1983 96:315-20.
Karsgaard AT. Spontaneous regression of retinoblastoma. A report of two cases. Can J Ophthalmol. 1971 6:218-22.
Boniuk M, Girard LJ. Spontaneous regression of bilateral retinoblastoma. Trans Am Acad Ophthalmol Otolaryngol. 1969 73:194-8.
Sanborn GE, Augsburger JJ, Shields JA. Spontaneous regression of bilateral retinoblastoma. Br J Ophthalmol. 1982 66:685-90.
Migdal C. Spontaneous regression of retinoblastoma in identical twins. Br J Ophthalmol. 1982 66:691-4.
Jakobiec FA, Tso MO, Zimmerman LE, Danis P. Retinoblastoma and intracranial malignancy. Cancer. 1977 39:2048-58.
Provenzale JM, Gururangan S, Klintworth G. Trilateral retinoblastoma: clinical and radiologic progression. AJR Am J Roentgenol. 2004 183:505-11.
Paulino AC. Trilateral retinoblastoma: is the location of the intracranial tumor important? Cancer. 1999 86:135-41.
Marcus DM, Brooks SE, Leff G, McCormick R, Thompson T, Anfinson S, Lasudry J, Albert DM. Trilateral retinoblastoma: insights into histogenesis and management. Surv Ophthalmol. 1998 43:59-70.
Whittle IR, McClellan K, Martin FJ, Johnston IH. Concurrent pineoblastoma and unilateral retinoblastoma: a forme fruste of trilateral retinoblastoma? Neurosurgery. 1985 17:500-5.
Mohney BG, Robertson DM, Schomberg PJ, Hodge DO. Second nonocular tumors in survivors of heritable retinoblastoma and prior radiation therapy. Am J Ophthalmol. 1998 126:269-77.
Moll AC, Imhof SM, Schouten-Van Meeteren AY, Kuik DJ, Hofman P, Boers M. Second primary tumors in hereditary retinoblastoma: a register-based study, 1945-1997: is there an age effect on radiation-related risk? Ophthalmology. 2001 108:1109-14.
Hausmann N, Stefani FH. Second nonocular tumors in cured unilateral retinoblastoma patients. J Cancer Res Clin Oncol. 1991 117:4-5.
Bhatnagar R, Vine AK. Diffuse infiltrating retinoblastoma. Ophthalmology. 1991 Nov;98(11):1657-61.
Materin MA, Shields CL, Shields JA, Eagle RC Jr. Diffuse infiltrating retinoblastoma simulating uveitis in a 7-year-old boy. Arch Ophthalmol. 2000 118:442-3.
Brisse HJ, Lumbroso L, Freneaux PC, Validire P, Doz FP, Quintana EJ, Berges O, Desjardins LC, Neuenschwander SG. Sonographic, CT, and MR imaging findings in diffuse infiltrative retinoblastoma: report of two cases with histologic comparison. AJNR Am J Neuroradiol. 2001 22:499-504.
Kao LY. Diffuse infiltrating retinoblastoma: an inherited case. Retina. 2000 20:217-9.
Perentes E, Herbort CP, Rubinstein LJ, Herman MM, Uffer S, Donoso LA, Collins VP. Immunohistochemical characterization of human retinoblastomas in situ with multiple markers. Am J Ophthalmol. 1987 103:647-58.
Donoso LA, Rorke LB, Shields JA, Augsburger JJ, Brownstein S, Lahoud S. S-antigen immunoreactivity in trilateral retinoblastoma. Am J Ophthalmol. 1987 103:57-62.
Donoso LA, Hamm H, Dietzschold B, Augsburger JJ, Shields JA, Arbizo V. Rhodopsin and retinoblastoma. A monoclonal antibody histopathologic study. Arch Ophthalmol. 1986 104:111-3.
Ohira A, Yamamoto M, Honda O, Ohnishi Y, Inomata H, Honda Y. Glial-, neuronal- and photoreceptor-specific cell markers in rosettes of retinoblastoma and retinal dysplasia. Curr Eye Res. 1994 13:799-804.
Margo C, Hidayat A, Kopelman J, Zimmerman LE. Retinocytoma. A benign variant of retinoblastoma. Arch Ophthalmol. 1983 101:1519-31.
Broughton WL, Zimmerman LE. A clinicopathologic study of 56 cases of intraocular medulloepitheliomas. Am J Ophthalmol. 1978 85:407-18.
Canning CR, McCartney AC, Hungerford J. Medulloepithelioma (diktyoma). Br J Ophthalmol. 1988 72:764-7.
Ulbright TM, Fulling KH, Helveston EM .Astrocytic tumors of the retina. Differentiation of sporadic tumors from phakomatosis-associated tumors. Arch Pathol Lab Med. 1984 108:160-3.
O'Brien JM, Marcus DM, Niffenegger AS, Bernards R, Carpenter JL, Windle JJ, Mellon P, Albert DM. Trilateral retinoblastoma in transgenic mice. Trans Am Ophthalmol Soc. 1989 87:301-22
Marcus DM, Lasudry JG, Carpenter JL, Windle J, Howes KA, al-Ubaidi MR, Baehr W, Overbeek PA, Font RL, Albert DM. Trilateral tumors in four different lines of transgenic mice expressing SV40 T-antigen. Invest Ophthalmol Vis Sci. 1996 37:392-6.
al-Ubaidi MR, Font RL, Quiambao AB, Keener MJ, Liou GI, Overbeek PA, Baehr W. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J Cell Biol. 1992 119:1681-7.
al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W. Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci U S A. 1992 89:1194-8.
Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992 9:955-66.
Hammang JP, Behringer RR, Baetge EE, Palmiter RD, Brinster RL, Messing A. Oncogene expression in retinal horizontal cells of transgenic mice results in a cascade of neurodegeneration. Neuron. 1993 10:1197-209.
Fung KM, Trojanowski JQ. Animal models of medulloblastomas and related primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1995 54:285-96.
Tamboli A, Podgor MJ, Horm JW. The incidence of retinoblastoma in the United States: 1974 through 1985. Arch Ophthalmol. 1990 108:128-32.