| A 12 year-old Girl with a
Retro-orbital mass. December, 2004, Case 412-1. Home Page |
Paula Aston B.A. (MS-IV)1, Kar-Ming Fung, M.D., Ph.D.2 Last updated November 30, 2004.
1 Fourth year medical student, Class of 2005, College of Medicine, University of Oklahoma and 2 Department of Pathology University of Oklahoma Health Science Center, Oklahoma City, Oklahoma
Clinical information: The patient was a 12 year-old girl who started to experience slowly progressive visual loss in the left eye 18 months ago. MRI demonstrated enlargement of the optic nerve by a fusiform tumor. The tumor is slightly hypointense on T1-weighed images and slightly hyperintense on T2-weighed images. There was no extension of the tumor beyond the orbit. The hypothalamus and optic chiasma were within normal limits. The left globe and the tumor was completely enucleated. At the time of surgery, the left eye was completely blind.
|
|
|
|
|
|
|
| A. | B. | C. | D. | E. | F. |
Pathology of the case:
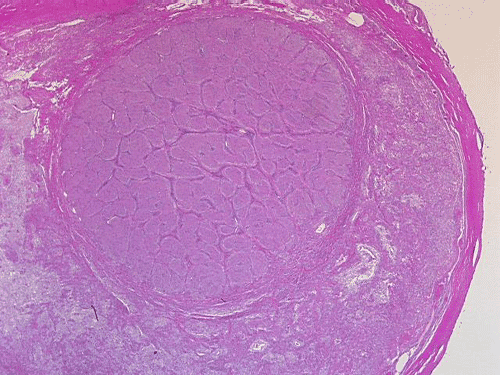
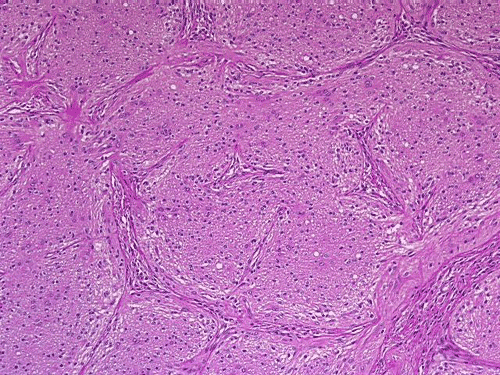
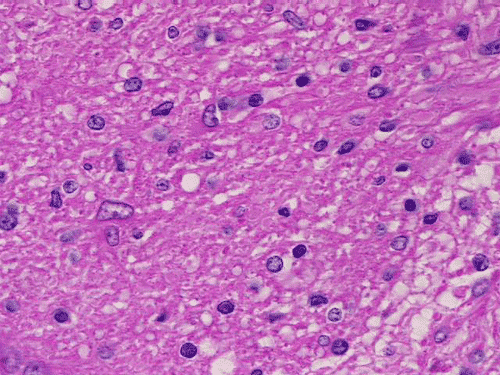
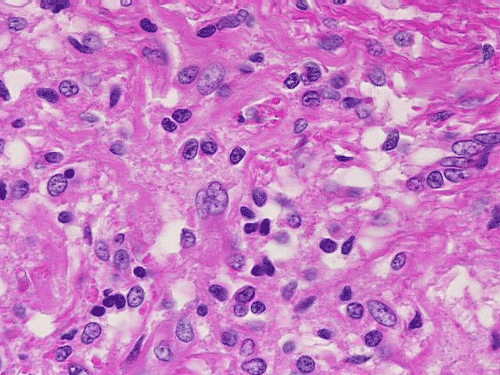
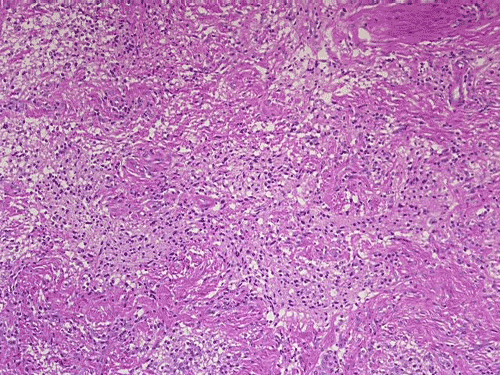
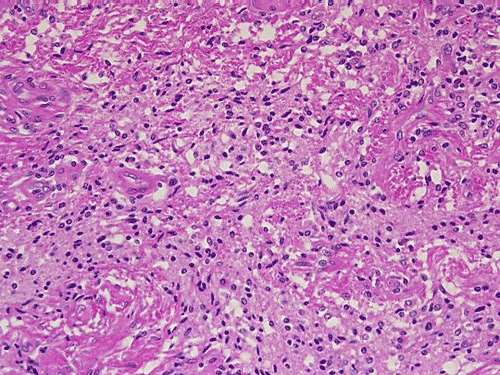
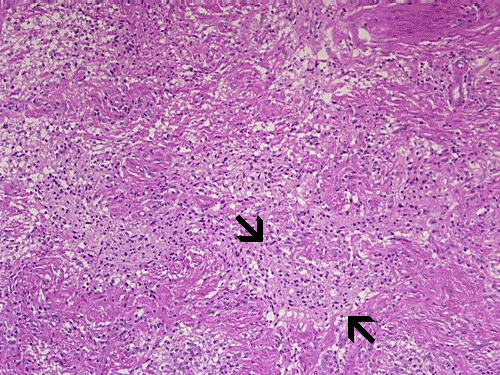
On low-magnification, cross section of the optic nerve gives a "double barrel" structure. The general outline of an optic nerve can still be appreciated (delimted by Þ in Panel A). There is a moderately cellular population of cells in between the dural sheath (… in Panel A) and the optic nerve. On medium-magnification, the vascular septa of the optic nerve is preserved (Panel B). On high-magnification, the optic nerve contains a mixture of cells with enlarged, mildly pleomorphic nuclei that are neoplastic astrocytes. Some cells with small nuclei are present and probably represent residual and reactive non-neoplastic astrocytes (Panel C and D). Cytoplasmic processes of some of tumor cells appear finely fibrillary (Panel C) while others are corase (Panel D). The cells in between the optic nerve and the dura is a mixture of irregular, hypercellular islands of neoplastic glial cells (delimted by Þ in Panel E) and hypocellular islands of slightly eosinophic, spindle cells. The later population represents reactive proliferation of meningothelial cells. The cytologic features are better appreciated in high-magnification (Panel F).
| DIAGNOSIS: Pilocytic astrocytoma of the optic nerve (optic nerve glioma). |
Discussion: General Information NF1 Clinical Imaging Pathology Differential diagnosis
General Information
Optic nerve glioma (ONG) refers to a group of glial tumors that are almost exclusively astrocytic neoplasms involving the anterior portion of the optic pathway. As a whole, ONGs make up 66% of all optic nerve tumor, 4% of orbital tumor and pseudotumor, 1% of all intracranial gliomas, and 3.6% of pediatric brain tumor 1, 2. About 90% of ONGs are found in patients under 20 years of age. There is a slight predominance of pediatric optic nerve gliomas in females with a male to female ratio of 2:3 3. In the adult population ONG are very rare. ONG is the most common tumor of the central nervous system NF1 with an incidence of about 15-40% in that population. Unilateral ONG in children is highly suspicious for NF1. The finding of bilateral optic nerve gliomas is pathognomonic for NF1.
ONG falls into two major clinical-pathological subgroups. The more common group involves children. These tumors are slowly progressive with many of them restricted to the optic nerve. However, there is also a significant number of visual pathway gliomas are isolated to the optic chiasma and retrochiasmal tracts, sparing the optic nerve.. When the tumor has no intracranial extension, the prognosis is excellent even when it is incompletely excised. When there is intracranial invasion, the prognosis is guarded. Most of these tumors have a histologic features of pilocytic astrocytomas. Spontaneous regression has been described in some of these tumors 4, 5, 6, 7. Very uncommonly, ONG in children can behave in a malignant manner 8.
ONGs occurring in adults are the much less common form. They occur in middle age patients and often cause extensive hypothalamic and even temporal lobe infiltration. Some of them may result in gross and radiographic appearance of gliomatosis cerebri. Often, they behave in a malignant manner. Histologically, they are often anaplastic astrocytomas or glioblastomas 9, 10, 11.
Neurofibromatosis 1 (NF1)
NF1 is an autosomal dominant disorder affecting about 1 in every 3000-4000 persons from all races and both genders. About half of the cases are sporadic. In familial cases, the expressivity of NF1 is extremely variable but the penetrance is 100%. No convincing genotype-phenotype correlations have been established. The NF1 gene was identified in 1990 on the long arm of chromosome 17q11.2and codes for the protein neurofibromin. NF1 is a pleiotropic congenital multiple dysplasia syndrome. The cardinal features of NF1 are multifocal hyperplasia and neoplasia in the supportive tissue throughout the entire nervous system. There is also a definite increased risk of systemic malignancies including chronic myelomonocytic leukemia (CML) (200 fold increased risk), neurofibrosarcoma, adenocarcinoma of the ampulla of Vater, melanoma, non-Hodgkin's lymphoma and lymphoblastic leukemia. Patients with NF1 are predisposed to develop malignant peripheral nerve sheath tumor (about 2-5% of NF1 patients will develop MPNST; risk of general population is 0.0001%) and these tumors often resulted from malignant transformation of pre-existing neurofibroma. Optic nerve gliomas is rare in the general population but common in NF1 patients. Other systemic manifestations such as café-au-lait macules, Lisch nodules (iris hamartomas), and osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortex with or without pseudoarthorsis and deformity of long bone are also present.
The clinical presentation of an optic nerve glioma in children is usually painless proptosis, nonpulsatile exophthalamus with decreased eye movement, and gradual, monocular vision loss occurring over a period of 6-12 months. The vision loss is sometime missed secondary to the child adjusting to it over time. Papilledema and optic nerve atrophy are common features on fundoscopic examination. Should the tumor expand to the chiasm, nystagmus may occur secondary to local compression and the visual deficit will likely be bilateral. Lesions extending into the hypothalamus can cause sleep and appetite changes and, if the lesion progresses to compress the third ventricle, obstructive hydrocephalus may result in a headache, nausea, and vomiting.
ONGs tend to grow outside the optic nerve and expand the subarachnoid and subdual space. ONGs are usually isointense to normal white matter on T1-weighted images and T2-weighed images. In contrast, CSF accumulation around the optic nerve is hyperintense on T2-weighted images and can therefore be well distinguished from ONG. As a result, the tumor is also well delimited by CSF on T2-weighted images. As gliomas of the optic pathway can arise as isolated chiasmal tumor and retrochiasmal tumors, evaluation of the entire optic nerve pathway is essential in patients with NF1.
Grossly ONG grows within the dural sheath to produce a fusiform enlargement. The neoplasm occasionally invades beyond the optic dural sheath.
Microscopically, ONG in children are almost all pilocytic astrocytomas. The optic nerve is expanded. The fibrovascular septa within the optic nerve are separated by the tumor cells but the structure can still be well recognized as an optic nerve on cross section. Three major patterns are recognized 1. In the first pattern, the tumor is finely reticulated. In the second pattern, the tumor has microscysts and is coarsely reticulated. In the third pattern, the tumor cells are coarsely fibrillated, spindle shaped, and form bundles. There is minimal pleomorphism in the nuclei and it is not always easy to separate the neoplastic cells from adjacent reactive gliosis. There is a usually lack in mitotic activity, endothelial proliferation, and necrosis. Immunohistochemistry, the tumor cells are strongly reactive for glial fibrillary acidic protein (GFAP).
ONGs often extend into the subarachnoid space and inflict a prominent proliferation and thickening of perioptic meninges. This proliferation is composed of meningothelial cells, fibroblasts, and neoplastic astrocytes. Such changes are also known as arachnoidal hyperplasia or arachnoidal gliomatosis. The tumor tends to have coarsely fibrillated spindle cells may not be easily to be separated from the meningothelial cells and fibroblasts due to the intermingled architecture. Immunohistochemistry for GFAP is very helpful in separating the two components.
Anaplastic astrocytomas and glioblastomas are found in adult patients and their histology is similar to tumors that arise within the brain and the prognosis is equally poor.
Differential diagnosis
Optic nerve choristoma 12, 13, 14 is a rare entity that can be easily mistaken as optic nerve glioma on clinical and radiographic examination. A choristoma is a mass of non-neoplastic, tumor-like masses of developmental origin in which they contain tissue foreign to the anatomical location. Optic nerve choristoma are composed of smooth muscle cells intermingled with dense fibrous connective tissue and atrophic optic nerve parenchyma. Histologically, therefore, is not difficult to be distinguished from ONG.
Optic nerve ganglioglioma 15, 16, 17 is a rare tumors that can cause diagnostic confusions. Involvement of the chiasm and hypothalamus is a common feature. They occur in young patients that range from 2 to 38 years of age which corresponds to the age of intracerebral gangliogliomas (Mclendon). Identification of the ganglionic component is the key to correct diagnosis.
Intraorbital meningiomas must also be considered in the differential diagnosis. Alternatively, ONG can be mistaken as meningioma clinically 18. Intraorbital meningiomas are far more common in women than men. They are most commonly seen in the third decade but has been described in other ages. About 40% of them occur in patients younger than 20 years of age and 25% occur in children younger than 10 years of age. Unlike the fusiform appearance of an ONG, the growth pattern of an optic nerve meningioma will fill the subarachnoid space along the length of the optic nerve giving it a railroad appearance on CT scan 19. Expansion of the optic nerve is often asymmetrical. On fundoscopic examination, there are chorioretinal striae and optocilliary shunt vessel are present on atrophic right optic nerve head. Histologically, they are usually of meningothelial type and mixed type. It should be noted that meninogthelial proliferation in areas invaded by the ONG is a common feature and should not be mistaken as meningioma. Keen observation, high index of suspicion, and clear knowledge of the location where the biopsy by communication with the neurosurgeon can help to avoid this trap.
Intraorbital schwannoma is rare. They usually involve the cranial nerve but not the optic nerve as the optic nerve is part of the CNS. On imaging studies, they may appear as peri-optic nerve mass or asymmetrical expansion of the optic nerve. Histologically, the spindle cells in schwannoma pose a diagnostic challenge on frozen sections as the astrocytes that extend into the subarachnoid space also tend to be spindle and coarsely fibrillary. On permenant sections, the distinction between these two entities should not be a major diagnostic challenge. Reticulin stain usually show deposition of reticulin substance around individual tumor cells. Schwannoma are also immunoreactive to GFAP but usually not to the same extent that is seen in ONG.
Reference:
Mclendon RE, Enterline DS, Tien RD, Thorstad WL, Bruner JM. Tumors of central neuroepithelial origin. In Russel and Rubinstein’s Pathology of Tumors of the Nervous System by Bigner DD, McLendon RE, Bruner JM. 6th edt., pg. 361-370, 1998, Arnold, London.
Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology. 2004 111:997-1008.
Hollander MD, FitzPatrick M, O'Connor SG, Flanders AE, Tartaglino LM. Optic gliomas. Radiol Clin North Am. 1999 37:59-71, ix.
Zizka J, Elias P, Jakubec J. Spontaneous regression of low-grade astrocytomas: an underrecognized condition? Eur Radiol. 2001 11:2638-40.
Yoshikawa G, Nagata K, Kawamoto S, Tsutsumi K. Remarkable regression of optic glioma in an infant. Case illustration. J Neurosurg. 2003 98:1134.
Parsa CF, Hoyt CS, Lesser RL, Weinstein JM, Strother CM, Muci-Mendoza R, Ramella M, Manor RS, Fletcher WA, Repka MX, Garrity JA, Ebner RN, Monteiro ML, McFadzean RM, Rubtsova IV, Hoyt WF. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001 119:516-29.
Schmandt SM, Packer RJ, Vezina LG, Jane J. Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg. 2000 32:132-6.
Safneck JR, Napier LB, Halliday WC. Malignant astrocytoma of the optic nerve in a child. Can J Neurol Sci. 1992 19:498-503.
Harper CG, Stewart-Wynne EG. Malignant optic gliomas in adults. Arch Neurol. 1978 35:731-5.
Wulc AE, Bergin DJ, Barnes D, Scaravilli F, Wright JE, McDonald WI. Orbital optic nerve glioma in adult life. Arch Ophthalmol. 1989 107:1013-6.
Millar WS, Tartaglino LM, Sergott RC, Friedman DP, Flanders AE. MR of malignant optic glioma of adulthood. Am J Neuroradiol 1995 16:1673-6.
Giannini C, Reynolds C, Leavitt JA, Schultz GA, Garrity JA, Ebersold MJ, Scheithauer BW, Salomao DR. Choristoma of the optic nerve: case report. Neurosurgery. 2002 50:1125-8.
Daxer A, Sailer U, Ettl A, Bleckenwegner G, Felber S. Choristoma of the optic nerve and chiasm. Arch Ophthalmol. 1992 110:236-8.
Zimmerman LE, Arkfeld DL, Schenken JB, Arkfeld DF, Maris PJ. A rare choristoma of the optic nerve and chiasm. Arch Ophthalmol. 1983 101:766-70.
Albayrak R, Albayram S, Port J, Degirmenci B, Acar M, Yucel A. Ganglioglioma of the right optic tract: case report and review of the literature. Magn Reson Imaging. 2004 22:1047-51.
Sadun F, Hinton DR, Sadun AA. Rapid growth of an optic nerve ganglioglioma in a patient with neurofibromatosis 1. Ophthalmology. 1996 103:794-9.
Lu WY, Goldman M, Young B, Davis DG. Optic nerve ganglioglioma. Case report. J Neurosurg. 1993 78:979-82.
Liauw L, Vielvoye GJ, de Keizer RJ, van Duinen SG. Optic nerve glioma mimicking an optic nerve meningioma. Clin Neurol Neurosurg. 1996 98:258-61.
Thiagalingam S, Flaherty M, Billson F, North K. Optic nerve glioma mimicking an optic nerve meningioma. Clin Neurol Neurosurg. 1996 98:258-61.
Cases of the Month Evaluation Coordinator: KarMing-Fung@ouhsc.edu
{kind=link}