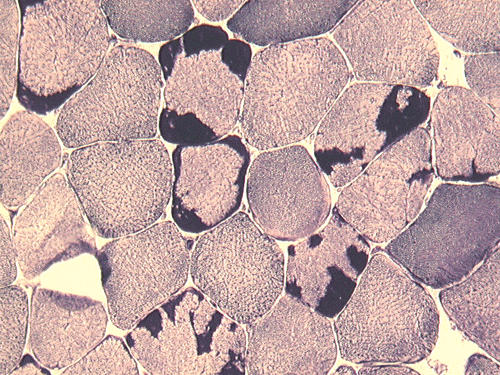
ATPase pH 9.4
NADH-TR
SDH
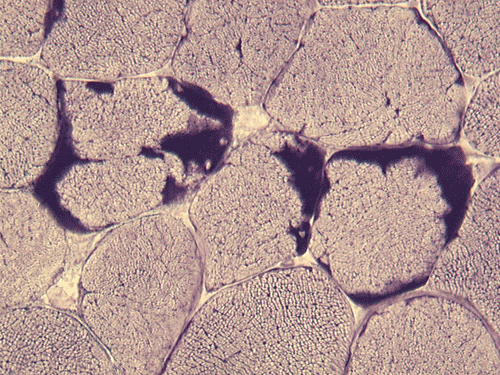
AD

| A 54 year-old Man with
Muscle Weakness and Decreased Endurance. June, 2005, Case 506-1. Home Page |
Waseem Ahmad, M.D. 1, Jiang Qian, M.D., Ph.D.2 Last update: August 1, 2005.
1 Department of Neurology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma and 2 Department of Pathology, Albany Medical College, Albany, NY
Clinical information: The patient was a 54 year old man with medical history of hypertension. He presented with ongoing fatigue and weakness with decreased endurance of exercise for several months. No episodic paralysis of any kind was experienced by the patient. He complained about headaches with blurred vision, weight loss and occasional muscle pain over the past 3 months. He was healthy-appearing and physical exam was unremarkable except for some tenderness to palpation of muscles. He did not smoke and only drank alcohol socially. Nine days prior to current visit, he had a rash on the left arm for which he was hospitalized. The culture of the rash grew a Streptococcus species. Laboratory evaluation showed an elevated CPK of 671 U/L (CK-mb was 16.6 U/L and percentage of CK-mb was 2.5%). A muscle biopsy was performed.
|
|
|
|
|
|
|
|
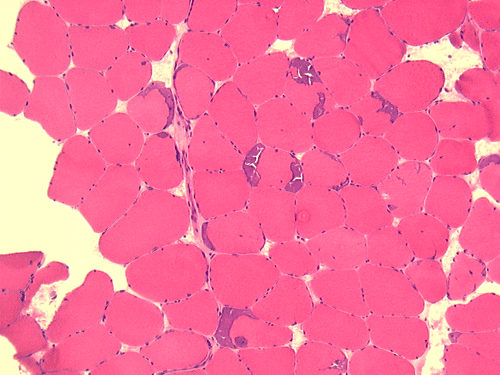
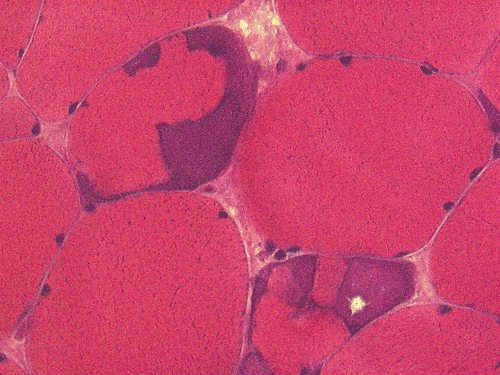
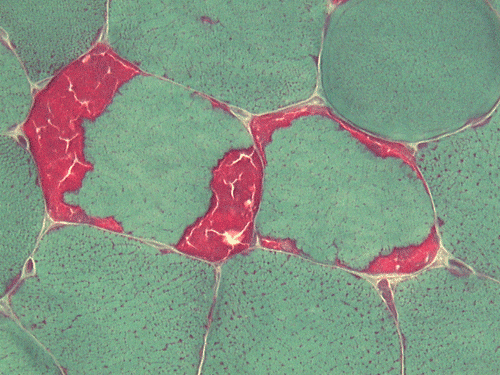
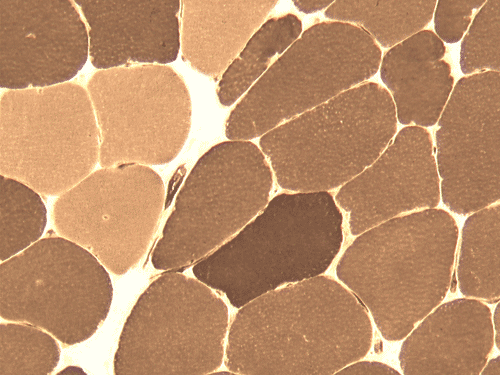
| A. | B. | C. |
D. ATPase pH 9.4 |
E. NADH-TR |
F. SDH |
G. AD |
|
|
|
|
|
|||
| H. | I. | J. | K. |
| HE | Hematoxylin-eosin stain. | SDH | Succinate dehydrogenase reaction. |
| MGT | Modified Gomori's srichrome stain. | AD | Adenylate deaminase |
| NADH-TR | NADH-tetrazolium reductase reaction. |
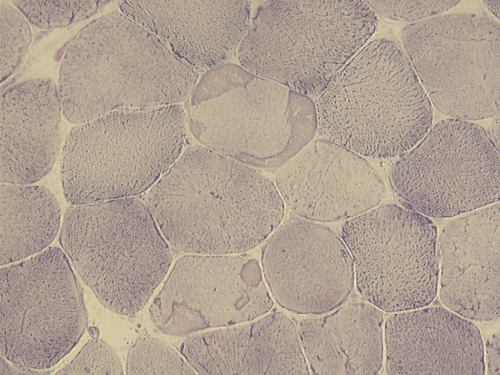
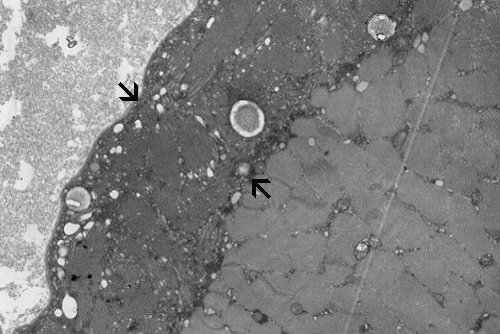
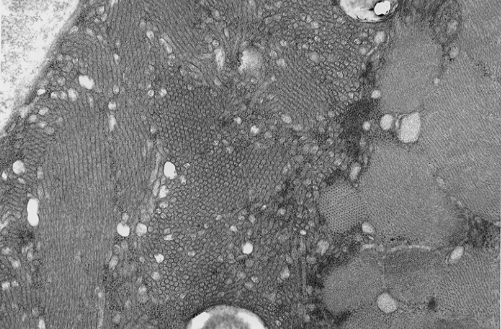
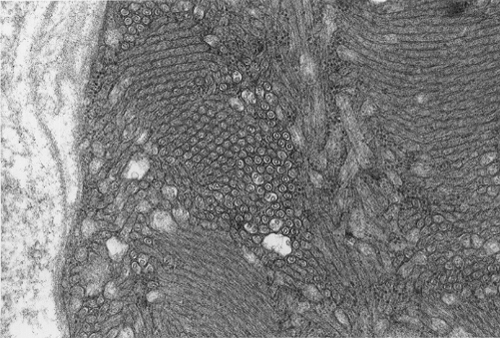
Pathology of the case: On hematoxylin-eosin stained frozen sections, there are many stongly basophilic, irregular subsarcolemmal depositions (Panel A and B). There is no significant variation in fiber diameter, increase in centrally located nucleus, inflammation, necrotic fiber, regenerating fibers or target fibers. These deposits appear bright red on modified Gomori's trichrome (Panel C). On ATPase reaction at pH 4.6 (dark fibers are type 1, pale fibers are type 2a while intermediate-intensity fibers are type 2b), there is no fiber type predominance (Panel D). Although the deposits are not visualized in the ATPase preparation, they are strongly reactive in NADH-TR reaction. Intesestingly, these deposits are not visualized in another preparation of oxidative enzyme- succinate dehydrogenase (Panel F). On the adenylate deaminase preparation, these deposits are strongly reactive (Panel G). The subsarcolemmal deposits are well visualized under the electron microscope which is electron dense (Panel H). On cross sections, these deposits are bundles of densely packed polygonal to round tubules with double walls that are arranged in small fascicles (Panel I and J). The tubules are about 50-80 nm in diameter. The tubular aggregates also arrange in longitudinal bundles that run along the myofibrils (Panel K).
| DIAGNOSIS: Tubular aggregate myopathy. |
Discussion: General Information Pathology Pathogenesis Differential diagnosis
General Information
Tubular aggregates are collections of small tubules at the electron microscopy level and as granular inclusions at the histochemical level. Tubular aggregates were first described by Engel in 1964 in periodic hypokalemic paralysis and in myotonic congenita and was initially believed to be abnormal mitochondria 1. Tubular aggregates do not contain succinate dehydrogenase but positive for lactate dehydrogenase and adenylate deaminase. These features argue against their mitochondrial origin. Tubular aggregates are often found in periodic paralysis and myotonic disorders. Tubular aggregates are also seen as inconstant feature or minor pathologic findings in a wide variety of myopathies and occasionally as incidental findings in normal skeletal muscles. In a study that included 1550 muscle biopsy specimens, tubular aggregates are found in 1% of the cases 2.
Tubular myopathy or myopathy with tubular aggregates refer to a rare entity that tubular aggregates represent the major, if not sole, pathologic changes of a primary myopathy that do not contain other diagnosable myopathic changes. No specific genetic alterations has been confirmed. Both sporadic and familial cases can occur. Both autosomal dominant and autosomal recessive pattern of inheritance have been described. In familial cases, both males and females can be affected.
Clinical manifestations of familial tubular myopathy fall into four known major groups 3. The first group are featured by slowly progressive, isolated, limb weakness with childhood or adult onset and without fatigability. The second type is featured by exercise induced muscle pain, cramps, and stiffness that can begin in childhood or adult and may be progressive and result in severe disability. Interestingly, muscle weakness is uncommon. The serum creatine kinase may rise after exercise. Although these features suggest a metabolic myopathy, the symptoms are not related to fasting and there is no myoglobinuria. In addition, the forearm ischemic exercise tests and serum electrolyte levels are normal. The third type are featured by myasthenic features, congenital myasthenic syndrome, limb weakness and easy fatigability in chilldhood or adulthood. The fourth type is the most unusual as it is not associated with obvious muscle symptoms. In contrast, this is a rare autosomal recessive disorder featured by slowly progressive visual loss with gyrate atrophy of the choroids and retina 3.
Tubular aggregates are well demonstrated in many routinely used histochemical stain. In general, tubular aggregates appear as irregular, subsarcolemmal depositions 4. They appear bright red in modified Gomori's trichrome. These aggregates appear strongly basophilic on hematoxylin-eosin stained frozen sections but they are quite indistinctive in paraffin embedded sections stained with hematoxylin-eosin. The aggregates appear intensely dark blue on NADH-TR reactions and this feature lead to the early impression that they are derived from the mitochondria. However, their lack of succinate dehydrogenase. Tubular aggregates are also strongly positive for lactate dehydrogenase and adenylate deaminase. These features argue against a mitochondrial origin. Tubular aggregates are usually but not invariably found in type II fibers.
On resin embedded semithin sections, tubular aggregates may measure over 40 mm in length on longitudinal sections. At the ultrastructural level, tubular aggregates appear as masses of bundles of long, straight, parallel tubules in between myofibrils especially beneath subsarcolemmal locations. They are often double barrel and contains an inner tube but they may be up to eight inner tubes. The outer tube is usually about 50 nm in diameter but may be as large as 80 nm. The inner tubules are 20-30 nm in diameter.
Other than tubular aggregate myopathy, the interesting fact that tubular aggregates have been described in multiple seemingly unrelated but well-defined neuromuscular diseases raises the question that formation of tubular aggregates may represent a shared response or reaction to pathologic insults or a common pathway in pathogenesis. The list of these myopathy is quite extensive and include, hypo-,hyper-,and normokalaemic periodic paralysis, thyrotoxic periodic paralysis, myotonia congenita, subclinical alcoholic myopathy, acromegaly, hyperaldosteronism, myasthenia gravis, neurogenic atrophy, malignant hyperthermia, inflammatory myopathy, whipples disease, porphyria cutanea tarda, as well as in otherwise normal individuals who have been exposed to large quantities of drugs. Other than familial cases, tubular aggregates are found only in males 3.
In a study of muscle fibers from affected members of a family with a dominantly inherited tubular aggregate myopathy showed that the presence of tubular aggregates potentiated the calcium loading capacity of slow muscle fibers, when compared with control individuals. Tubular aggregates were functional equivalents to hypertrophy of the terminal cisterns of the sarcoplasmic reticulum and represented an adaptive response to an increased calcium influx. In electron microscopy, there are suggestive connections between tubular aggregates and lateral cisternae of sarcoplasmic reticulum 5,6,7. Dilated sacs of sarcoplasmic reticulum may be associated with the aggregates.
Abnormalities of intracellular Ca++ regulation, may also apply to other disorders known to be associated with tubular aggregates in skeletal muscle, particularly those caused by mutations in muscle-specific channel genes. This hypothesis concurs with the observation that tubular aggregates are most often found in periodic paralysis. The products of these genes, which are located at the neuromuscular junction, in the muscle fiber membrane and T-tubular system, or in the sarcoplasmic reticulum itself, are key players in the complex processes of muscle fiber excitation and excitation-contraction coupling, and when defective, are likely to have significant effects on intracellular Ca++ homeostasis. Perturbations in Ca++ flux regulation, possibly caused by a local shortage of energy donors, may also account for tubular aggregate formation in OAT (ornithine aminotransferase) deficiency. On the basis of available animal models, it is proposed that tubular aggregate formation in skeletal muscle occurs in response to genetic and/ or functional abnormalities, affecting specific components of the machinery involved in muscle fiber excitation, excitation-contraction coupling, or intracellular Ca++ flux regulation 5,6,7.
Reference: