Trichrome
Congo red
Congo red
Congo red
polarized light
| A 54 year-old Man with a
Colonic Mass and Blood in Stool. February, 2006, Case 6012-2. Home Page |
George Pirumyan, M.D., William F. Kern, M.D., Barbara Bane, M.D. Last update: March 30, 2006.
Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma.
Clinical information: The patient was a 54 year-old who was admitted for evaluation of dysphagia and heartburn with lower gastrointestinal symptoms that included bright-red blood per rectum, mixed with the stool. On physical examination, the patient appeared to be well built and well nourished with no significant findings. On further investigation, he was found to have a mass in the region of the sigmoid colon. Colonoscopy revealed a nearly circumferential large lesion at 30 cm away from the anal verge. The mucosa covering the mass as well as from other part of cecum and colon was otherwise unremarkable. The mass was excised by a sigmoid colectomy.
Grossly, surgical specimen consists of segment of colon, measuring 7.0 cm in length and 6.0 cm in circumference. Mucosal surface reveals a tan, raised, polypoid lesion (4.5 x 3.5 x 2.0 cm), lesion extends into submucosal layer up to 1.5 cm.
The followings are representative microphotographs of the sigmoid colon:
|
|
|
|
|
|
|
|
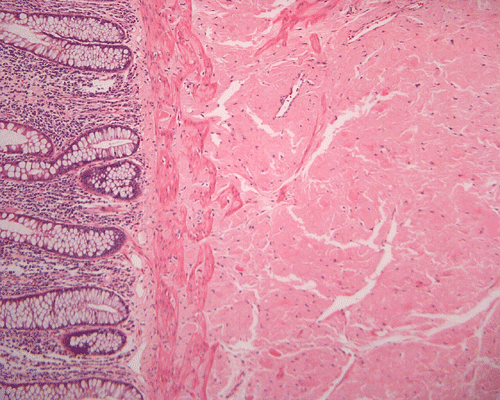
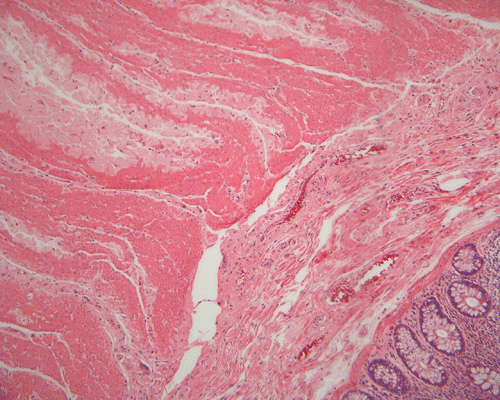
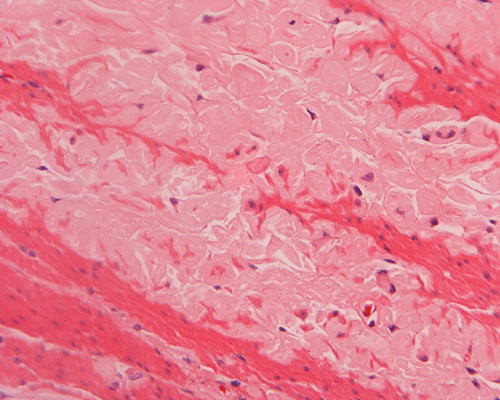
| A. | B. | C. |
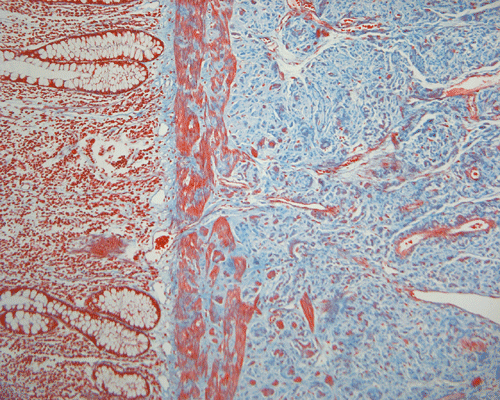
D. Trichrome |
E. Congo red |
F. Congo red |
G. Congo red polarized light |
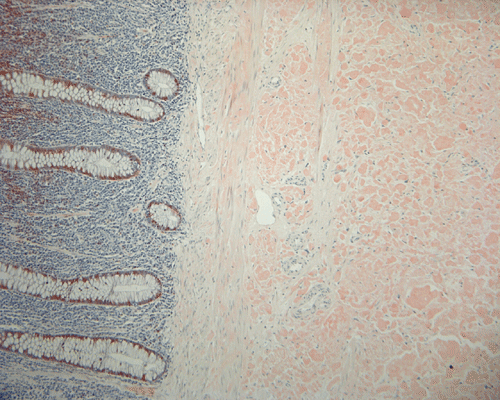
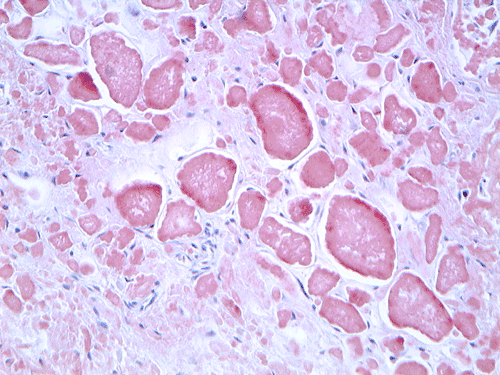
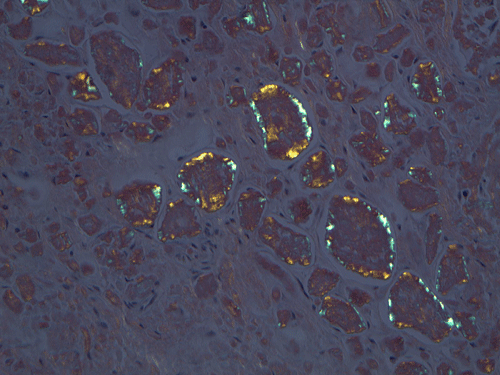
On low magnification, the mucosa appear intact and unremarkable. However, the submosa and muscular layer is replaced by a pale eosinophilic, hypocellular, amorphous material (Panel A). These material separate the smooth muscle bundles (Panel B and C). On Masson's trichrome, these material appears blue (Panel D). They are negative for elastic stain (Panel E). These material are stained orange by Congo Red stain (Panel F and G) which gives an apple green birefrigence on polarized light (Panel H). Electron microscopy was performed and revealed non-branhing straight fibrils ranging from 6.6 to 8.3 nm in diameter consistent with amyloid.
Immunohistochemistry was performed and showed strong reactivity for lambda light chain. Numerous plasma cells within the mucosa overlying the amyloid deposition are also postive for lambda light chains. The results suggest a monoclonal lambda light chain plasma cell dyscrasia but it could not be further classified in the excised specimen.
Aftermath:
No protein was revealed in his urine. Theperipheral blood count was negative. Additional clinical work up did not revealed any evidence of a lymphoproliferative disorder. The patient was followed for at almost 5 year when this case report was prepared. Other than coronary heart disease, diabetes, hypertension, hyperlipidemia, and the resection of a lipoma, there was neither a recurrence of his amyloid tumor or development of plasmacytoma or other lymphoproliferative disorders that would explain his amyloid deposition.
| DIAGNOSIS: Amyloidoma of the colon. |
The term amyloid was introduced in 1854 by the German physician scientist Rudolph Virchow. The name amyloid was derived from the Latin amylum and the Greek amylon 1. When Virchow, looking for cellulose-like materials using iodine, first transferred the designation ‘amyloid’ from plant biology to a human structure, he did so to a structure in the brain called the corpora amylacea 2.
Amyloid protein is defined by its resistance to proteolysis and its three-dimensional configuration as a beta pleated sheet. Amyloidosis results from tissue deposition of amyloid protein that leads to deterioration of function of the affected organ or tissue. In essence, amyloidosis belongs to a class of disease which share the features of change in conformation of protein structure as a common biochemical feature. Other examples of diseases featured by distorted protein conformation include sickle cell disease and prion encephalopathies. Change in conformation of a diverse group of proteins including immunoglobulin and transthyrectin can also lead to amyloid deposition.
The modern nomenclature of amyloidosis now includes 25 human and 8 animal fibril proteins 3. There are several subtypes of amyloidosis including primary amyloidosis, also known as light chain amyloidosis, secondary and familial amyloidosis. All forms are designated “A” plus a suffix that denotes the precursor protein 3. Currently amyloid is defined as an extracellular deposit of protein fibrils with this organization, which gives the characteristic apple green birefringence when stained with Congo red and examined under polarized light 4.
Classifications of amyloidosis have grown from the simple notion of primary and secondary amyloidosis, based on the broad concepts of association with myeloma (primary) or chronic illnesses (secondary), or concepts of “systemic” or “localized” to current designations that reflect the precursor protein 4. It is very important to determine the type of amyloid because the prognoses for the 3 forms of systemic amyloid and the treatment of the 3 disorders are vastly different. One often involves chemotherapy and stem cell transplantation; another might lead to liver transplantation. Accurate classification becomes imperative 4. The two major types of systemic amyloidosis are AL (primary amyloidosis or myeloma-associated amyloidosis) and AA (secondary or reactive amyloid).
In the systemic or generalized form of amyloidosis, deposits of amyloid are found in many organs. It can involve vital organs, such as kidneys, heart, liver and rarely brain. In contrast localized forms occur in one single tissue or organ. Localized amyloid deposits have been reported virtually in every organ, including amyloidosis of tongue, seminal vesicles and vasa deferentia, stomach, penis, skin 5, 6, 7, 8, 9. Only 2 articles were found describing localized amyloidosis of sigmoid colon in absence of systemic amyloidosis 10, 11.
Amyloid deposition can also occur in the peripheral nerve and lead to peripheral neuropathy. Primary cerebral amyloid angiopathy is typically associated with deposition of amyloid b (Ab) which is a product of cleavage of the amyloid precursor protein (APP). Primary cerebral amyloid angiopathy is not uncommon and manifestations occur predominantly in older patients. Repeated hemorrhages are common in these patients. While most of the cases are sporadic, familial cases have been identified and are often associated with mutation of the APP protein. Ab protein deposition is typically found in plaques and blood vessels in Alzheimer’s disease 12, 13.
The incidence of amyloidosis is 8 patients per million per year 4. Patients who are on chronic dialysis are at risk for developing amyloidosis. The two most common forms of systemic amyloidosis are light-chain (AL) amyloidosis, with an incidence of approximately 1 case per 100,000 person-years in Western countries, and reactive amyloidosis due to chronic inflammatory diseases (e.g., rheumatoid arthritis and chronic infections) 1. On the contrary, AA amyloidosis, related to familial Mediterranean fever, is associated with certain ethnic groups: Sephardic Jews (37%) and Armenians living in Armenia (24%), and less frequently in Ashkenazi Jews and Arabs (8% to12%) 14.
Multiple mechanisms of amyloid formation have been proposed 14. Briefly, these are:
Histologic diagnosis
Amyloid can be detected on routine hematoxylin and eosin (H&E) slides, all forms of amyloid deposits are amorphous and homogenous, with pale eosinophilic areas 15. Microscopic characterization of amyloid by special studies is necessary to confirm diagnosis of amyloidosis. Congo red histochemical stain remains gold standard for diagnosis and characterize by common to all amyloid aggregates orange or red color on light microscopy and clear apple-green birefringence under polarised light 1, 2. Most of the other stains available are not recommended to be used alone and are listed with decreasing specificity and sensitivity: Sirius red, thioflavine T, thioflavine S, toluidine blue, p-dimethylaminobenzaldehyde-nitrite, alcian blue, and crystal violet 15. Electron microscopy studies indicate that the basic assembled unit is a thin filament of about 4 nm in diameter, and that the mature fibrils consist of a few such proto-filaments aligned in parallel, often with a twist 2. Some other authors describe amyloid filaments under electron microscope as loose meshwork of 7–10 nm wide rigid, non-branching, hollow fibrils of indeterminate length. The fibrils measure from 30 to more than 1000 nm in length and are usually found in aggregates in extracellular spaces 15, 16.
Immunohistochemistry can be helpful in elucidating the nature of amyloid deposition. The more common protein that can be detected in amyloidosis are immunoglobin, transthyrectin, and Ab. Amyloid often co-deposits with P-component. Although non-diagnostic, demonstration of P-component by immunohistochemistry is a sensitive way to detect amyloid and is expecially helpful when the deposition is small, such as those in amyloid peripheral neuropathy. In vivo diagnosis of amyloidosis has also been achieved by 123I-labeled serum amyloid P component scintigraphy 17.
Gastrointestinal tract is one of the regions to be commonly involved in the systemic amyloidosis. However, amyloidosis confined to the sigmoid colon is a rare occurrence. In excised specimen similar to the one being described here, diagnosis of amyloidosis should not be a problem. In small endoscopic biopsies, the detection rest heavily on astute observation and high index of suspicion of the pathologist. Knowledge of clinical history that may produce amyloidosis is helpful. Special stains should be performed when amyloid deposition is suspected.
Reference: