| A 45 year-old Man with New
Onset Seizure and an Enhancing Mass in the Brain. May, 2007, Case 705-1. Home Page |
Katerine Seywerd, M.D.1, Min Wang, M.D., Ph.D.2 Last update: Decemb er 1, 2007.
1 Department of Pathology, University of Oklahoma Health Science Center, Oklahoma City, Oklahoma, 2 Department of Pathology, University of Texas, Houston, Texas
Clinical information: The patient was a 45 year-old man who complained of headache and two attach of seizures in the past four months. On MTI, there was a 3 cm enhancing, plaque-like mass in the parietal lobe. It was not certain whether the mass was extra-axial or intra-axial but meningioima was suspected. No other abnormal findings were noted on systemic work up. The mass appeared tightly adhered to the underlying brain parenchyma upon surgical resection. The following images were obtained from the excised specimen
|
|
|
|
|
|
|
| A. | B. | C. | D. | E. | F. |
Pathology:
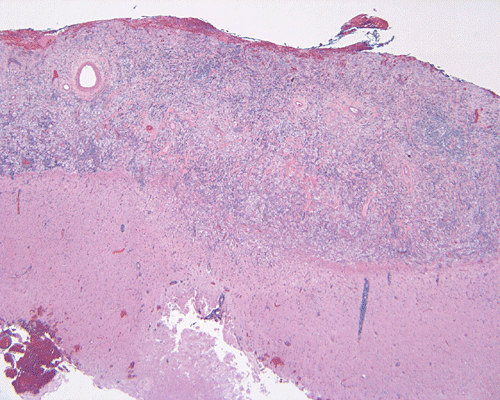
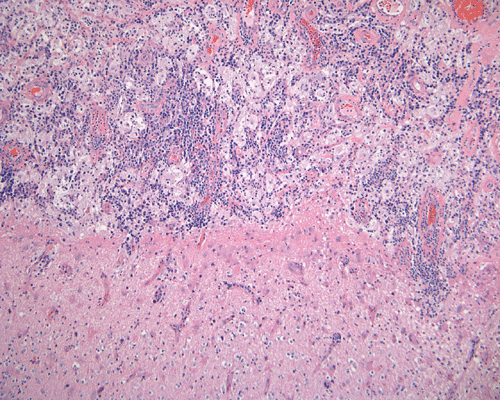
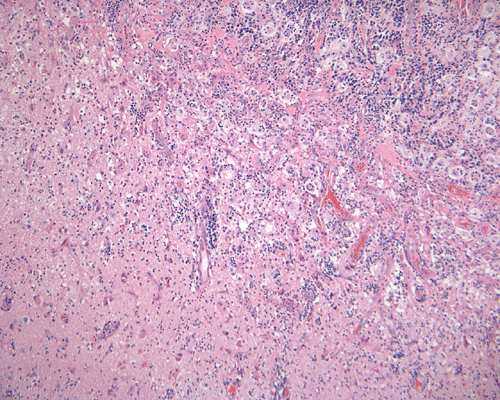
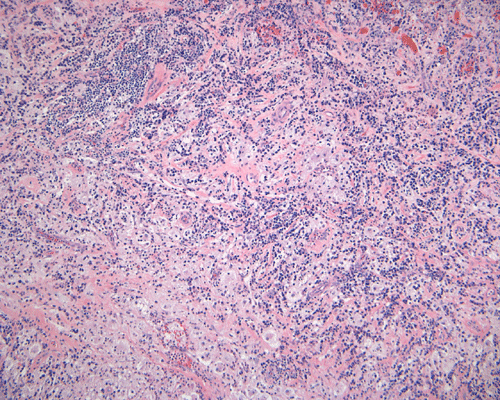
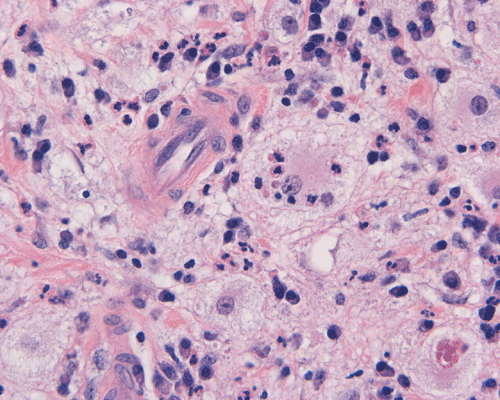
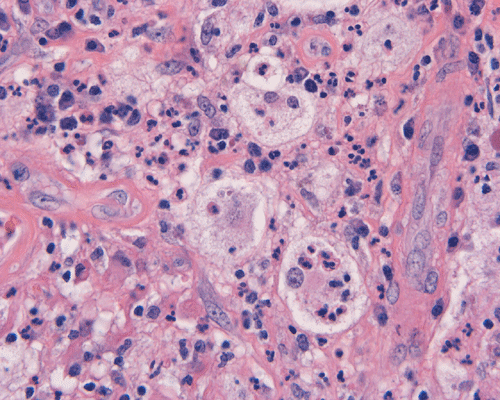
The specimen is in the form of a plaque-like, firm lesion adhered to a layer of brain parenchyma on one side (Panel A) The lesion infiltrate into the underlying brain parenchyma which leads to the formation of an irregular interphase. There is also perivascular accumulation of lymphocytes around blood vessels (Panel B and C). On low-mangification, the lesion appears to be a mixture of lymphocytes and large cells with foamy histiocytes. The salient feature of the lesion is that of a mixture of mature lymphocytes with large to giant histiocytes (Panel D). The histopathologic picture is rather homogeneous in different parts of the lesion. On high-magnification, the large to giant histiocytes has enlarged nuclei and many of them contain engulfed lymphocytes or polymorphonuclear leukocytes (emperipolesis ) (arrows in Panel E an F).
| DIAGNOSIS: Sinus histioicytosis with massive lymphadenopathy (Rosai-Dorfman disease). |
Discussion:
General Information:
In 1969, Rosai and Dorfman described a benign histocytic proliferative disorder which they subsequently named sinus histiocytosis with massive lymphadenopathy (SHML). Albeit, this entity is also popularly known as Rosai Dorfman diease (RDD) 1.
RDD is seen world wide among all races and ethnic groups. Males are more commonly affected than females. Although it often presents as lymph node enlargement and suggests a malignant neoplasm of the hematopoietic system, RDD is an uncommon benign, non-neoplastic and often protracted histiocytic proliferative lesion with systemic symptoms. RDD is usually a self-limited dsease. The potentially alarming clinical presentation often appears in the 1st or 2nd decade of life as a febrile illness that is accompanied by extensive but painless lymphadenopathy (most commonly cervical), fever, anemia, an elevated erythrocyte sedimentation rate, neutrophilia, and polyclonal hypergammaglobulinemia. Unusual cases have also been described in patients in their 8th decades. Isolated intracranial or brain involvement is rare but well documented 2, 3, 4, 5, 6. A few cases with isolated involvement of the spinal cord have also been reported 2.
The majority of patients do not require treatment, demonstrating spontaneous resolution or stable disease. In a case study of 11 patients with isolated central nervous system (CNS) involvement by Andriko et al 2 a surgical approach was taken. No chemo- or radiotherapy was utilized. With complete local excision no recurrence was noted after 15 months. In those with subtotal resection no progression was observed, clinically or radiographically. However, in some cases the disorder runs a progressive course and/or threatens vital structures and functions, which dictates some form of intervention. It is not clear what constitutes the best approach for those cases. Jabali 7 reported success in managing an isolated case with combination chemotherapy.
The etiology of RDD is unknown although a viral pathogenesis has been postulated. Numerous reports have identified human herpesvirus 6 (HHV-6) in visceral and cutaneous lesions 8. However, HHV-6 has frequently been found in many reactive and infectious disorders of lymphoid tissue, and its presence in Rosai-Dorfman disease is non-specific. Immunophenotypic profiling and studies of monokine expression suggest an origin from activated macrophages, which have been shown to produce IL-1-beta and and TNF-alpha. Molecular studies using polymorphic regions of the human androgen receptor locus have demonstrated that RDD is a polyclonal disorder 2.Extranodal involvement:
Extranodal involvement, either in isolation or in association with lymphnodes, can be seen in 25 -40 % of cases, with skin, orbit and upper respiratory tract being the most common sites 9. It may also involve the paranasal sinuses, soft tissue, bone, and less commonly salivary gland, oral cavity, kidney, lower respiratory tract, larynx, and, rarely, other locations such as the CNS. Immune disorders occur in approximately 13 percent of patients, manifesting most frequently as anti-erythrocyte autoantibodies and joint disease 10.
Extranodal RDD with CNS involvement is observed in fewer than 5% of all patients. Involvement of the CNS is not considered a sign of aggressive behavior. In contrast to nodal based RDD, RDD of the CNS often occurs in older patients, usually the 4th and 5th decade 2. Two-thirds of such patients have systemic involvement, especially the skeletal system and salivary glands. Intracranial lesions without associated lymphadenopathy or systemic involvement (primary RDD of the CNS) are quite rare. The most common clinical presentations include headaches, seizures, numbness or paraplegia. Radiographically single or multifocal dural based lesions are seen on imaging. This often leads to an initial clinical and radiological impression of meningioma 2. Uncommonly, isolated parenchymal involvement without dural involvement occurs. Intraparenchymal lesions are almost always solitary lesions and are much less common.
Pathology:
Grossly, dural based RDD are firm, often plaque like lesions that resemble meningioma but they are firmly adhered to the brain.
Histiologically, the features of cranial RDD are similar to those occurring in the lymphnodes. Lesions are composed of sheets or syncytia of large, pale histiocytic cells with abundant granular eosinophilic cytoplasm and ill-defined or feathery cytoplasmic borders in a lymphocytic, chronic inflammatory cell background. Extranodal sites commonly exhibit a degree fibrosis and this can obscure the visibility of the histiocytic proliferation. Nuclei of the large histiocytes vary in size and may be large, round and vesicular with smooth nuclear contours 2. A mild degree of cytological atypia is commonly seen. Most nuclei contain a single small nucleolus, but others possess multiple nucleoli or a single prominent nucleolus. Mitotic figures are usually difficult to detect or are absent altogether. The prominent background of chronic inflammation is largely comprised of a mature plasmacytic infiltrate with interspersed lymphoid follicles 2. Emperipolesis is consistently found in nodal disease and often is less evident at extranodal sites. Its recognition is key to correct diagnosis.
In all cases the histiocytes of RDD are storongly immunoreactive for S100 protein. Immunostaining with S-100 can facilitate histiocyte identification 2. Other markers are often positive and include CD68, HAM-56, and MAC-387. Histiocytes in RDD also express CD11c, CD14, and CD33, as well as lysozyme, α1-antitrypsin, and α1-antichymotrypsin. Half of the cases express CD30. Pancytokeratin immunostain (AE1/3), CAM 5.2, and CD31 are negative 10. RDD are negative for CD1a and no Birbeck granules are not seen with electron microscopy. The last two features help to differentiate these lesions from Langerhans cell histiocytosis 11.
Differential Diagnosis:
Histologic differential diagnostic considerations include several hematopoietic and primary CNS lesions. Primaries among these are lymphoplasmacytic-rich meningioma, Langerhans cell histiocytosis, hemophagocytic lymphohistiocytosis, lymphoma, post-transplantation lymphoproliferative disorder, plasma cell granuloma and granulomatous inflammation (ie- TB and sarcoid).
Lymphoplasmacytic meningioma can be challenging as it elicits a pronounced chronic inflammatory reaction, which may be confused with the infiltrates of RDD-CNS 2. Recognition of typical meningioma histology coupled with confirmatory immunostaining for epithelial membrane antigen (EMA) readily differentiates the two processes. It is also worth noting that meningiomas are variably positive for S-100 and the positive cells should not be interpreted as RDD associated histocytes.
Primary Langerhans cell histiocytosis in the CNS is rare and usually arises along the hypothalamic pituitary axis as discrete nodules attached to the dura 2. Occurrence as a localized osteolytic lesion in the skull of a child or young adult is a far more common presentation. Secondary involvement of the CNS and skull by systemic Langerhans cell histiocytosis can occur. It may exhibit a xanthomatous appearance microscopically 2. Cytologically, the nuclei of Rosai-Dorfman histiocytes lack lobulation and longitudinal nuclear grooves (ie- coffee bean nuclei) characteristic of Langerhans cells. Eosinophils are often prominent in Langerhans histiocytoses and are rarely observed in the cellular infiltrates of RDD. Both RDD and Langerhans' cell histiocytosis are immunoreactive with S100, but CD1a is typically negative in RDD 2. Unlike Langerhans cell histiocytosis, Birbeck granules are not seen with electron microscopy in RDD 7.
The prominent fibrosis seen in RDD of the CNS may have a distinctly nodular appearance suggestive of the nodular sclerosing variant of Hodgkin's disease. A key point to remember is that Hodgkin's disease in the CNS is extremely rare and will typically be associated with relapse 2. Although classic Reed Sternberg cells are readily distinguished from RDD histiocytes, mononuclear variants, particularly lacunar cells may mimic RDD histiocytes. Classic Reed Sternberg cells and variants, however, lack emperipolesis and are S-100 negative. Reed Sternberg cells are typically positive for CD15 and CD30.
RDD and primary lymphoma of the CNS share the common feature of perivascular lymphocytic cuffs. Some cases of RDD may be lymphocyte-rich and emperipolesis may not be easily identified. However, most primary lymphomas of the CNS are high grade tumor with diffuse large-B cell lymphoma as the most commonly seen entity. RDD would not have the same high level of pleomorphism. Primary lymphomas of low histologic grade do occur in CNS but they are rare. Differentiation of these tumors from RDD can create difficulties. Lymphomas exhibit malignant cytologic features (especially the aggressive variants) and predominance of one cell type, usually B-cell, can be demonstrated by immunohistochemistry. Lymphocytes in RDD are bland, polyclonal, and show a mixture of B- and T-cell phenotypes. Multiple myeloma and plasmacytoma are characterized by monoclonal proliferation of tumor cells 2.
Reference:
Rosai J, Dorfman RF..Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol1969 87 :63 –70.
Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol. 2001 Mar;14:172-8.
Toh CH, Chen YL, Wong HF, Wei KC, Ng SH, Wan YL. Rosai-Dorfman disease with dural sinus invasion. Report of two cases. J Neurosurg. 2005 102:550-4.
Griffiths SJ, Tang W, Parameswaran R, Kelsey A, West CG. Isolated intracranial Rosai-Dorfman disease mimicking meningioma in a child. Br J Neurosurg. 2004 18:293-7.
Konishi E, Ibayashi N, Yamamoto S, Scheithauer BW. Isolated intracranial Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy). AJNR Am J Neuroradiol. 2003 24:515-8.
Sundaram C, Uppin SG, Prasad BC, Sahu BP, Devi MU, Prasad VS, Purohit AK. Isolated Rosai Dorfman disease of the central nervous system presenting as dural-based and intraparenchymal lesions. Clin Neuropathol. 2005 24:112-7.
Jabali Y, Smrcka V, Pradna J. Rosai-Dorfman disease: successful long-term results by combination chemotherapy with prednisone, 6-mercaptopurine, methotrexate, and vinblastine: a case report. Int J Surg Pathol. 2005 13:285-9.
Levine PH, Jahan N, Murari P, Manak M, Jaffe ES. Detection of human herpesvirus 6 in tissues involved by sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Infect Dis. 1992 166:291-5.
Sennes L, Koishi H, Cahali R, Sperandio F, Butugan O. Rosai-Dorfman disease with extranodal manifestation in the head. Ear Nose Throat J. 2004 83:844-7.
Paulli M, Rosso R, Kindl S, Boveri E, Marocolo D, Chioda C, Agostini C, Magrini U, Facchetti F. Immunophenotypic characterization of the cell infiltrate in five cases of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Hum Pathol. 1992 23:647-54.