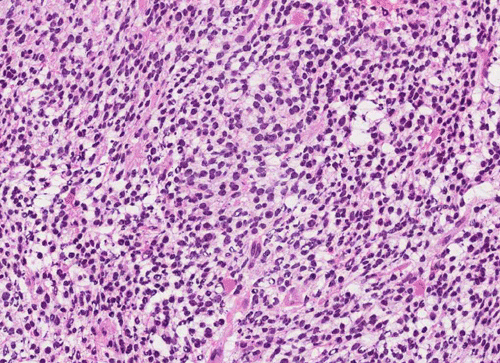
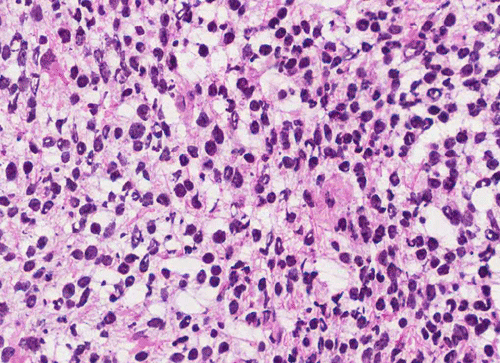
T1+C
T2
Squash
Squash
Frozen
Frozen
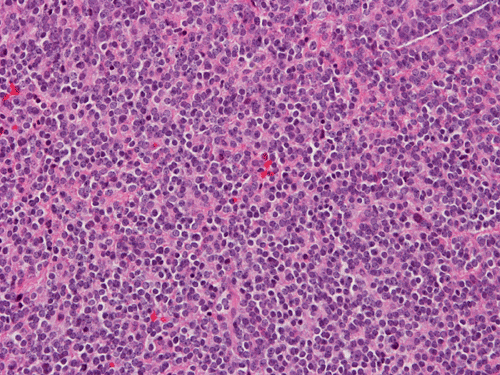
Ki67
Neurofilament
GFAP
| A 49 year-old Man with
Headache and a Pineal Mass. January, 2009, Case 901-1. Home Page |
Catherine E. Porter, B.S. (MS-IV), Kar-Ming Fung, M.D., Ph.D. Last update: April 10, 2009.
1 Fourth year medical student, Class of 2010, College of Medicine, University of Oklahoma, Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
The patient was a 49 year-old Caucasian man who presented with a six week history of headaches and complaint of “pressure” in his head. Despite attempted treatment with antibiotics for suspected sinusitis, the patient’s symptoms of pain and pressure worsened significantly during the fifth and sixth weeks. A CT scan performed in an outside hospital was reported to be negative for sinusitis. The patient subsequently presented to the neurosurgical service of this hospital.
His chief complaint was headache. He denied any visual changes, nausea, vomiting, focal weakness or paresthesias. Physical exam was unremarkable with no significant abnormal findings. On neurologic exam, the patient was found to be neurologically intact with no frank papilledema and no focal findings other than bilateral optic disc margin elevation. The patient was married with a past medical history of interstitial cystitis. He denied any use of tobacco or illicit drugs, any excessive alcohol consumption and any recent travel.
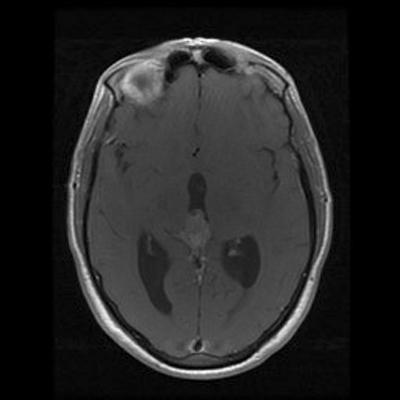
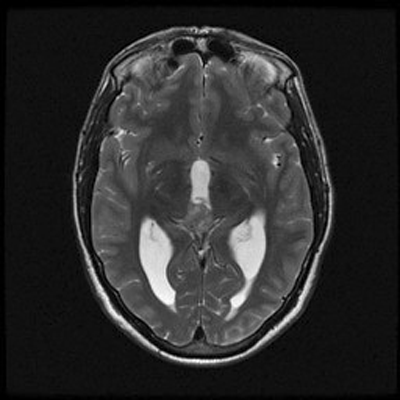
An MRI of the brain with and without contrast revealed a 2.6 x 1.3 x 1.6 cm (AP x CC X W) heterogenous lesion within the region of the pineal gland and projecting into the third ventricle. The lesion had both solid and cystic components and demonstrated heterogenous enhancement on T1-weighed images. The enhancement is located medial to the veins of Rosenthal, inferior to the cerebral veins, and posterior to the vein of Galen. The mass is isointense to the cortex on T2-weighed images. No significant midline shift or significant edema was observed. The ventricular system was prominent in size, consistent with mild hydrocephalus. Basilar cisterns were preserved and normal cerebrovascular flow-voids were seen. No abnormalities of the cerebellar or cerebral parenchyma, the brainstem or the cervical spinal cord were observed. Dural venous sinuses and meninges were unremarkable. Regions of the orbit and sella were unremarkable. Paranasal sinuses demonstrated paranasal sinus disease of the bilateral maxillary and left ethmoidal sinuses.
The patient was placed on dexamethasone and a third ventriculostomy with endoscopic biopsy of the pineal mass was performed. As per the neurosurgeon, the tumor appeared pedunculated. The following are representative images.
|
|
|
|
|
|
|
|
|
A. T1+C |
B. T2 |
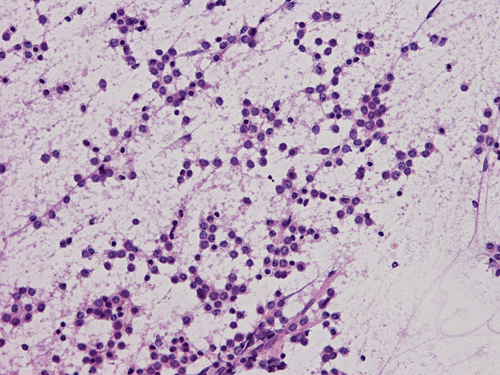
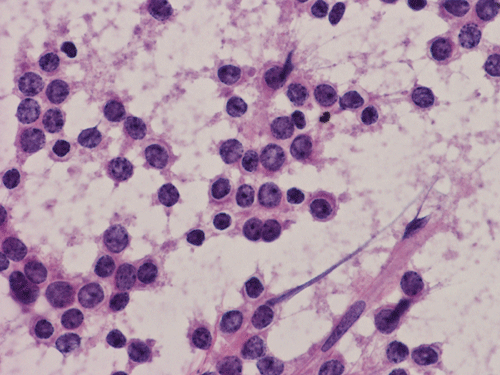
C. Squash |
D. Squash |
E. Frozen |
F. Frozen |
G. |
|
|
|
|
|
|
|
|
| H. | I. | J. |
K. Ki67 |
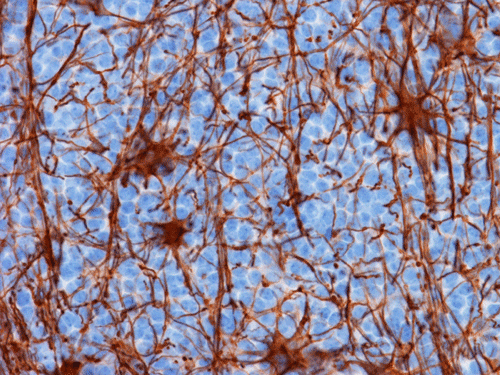
L. Neurofilament |
M. GFAP |
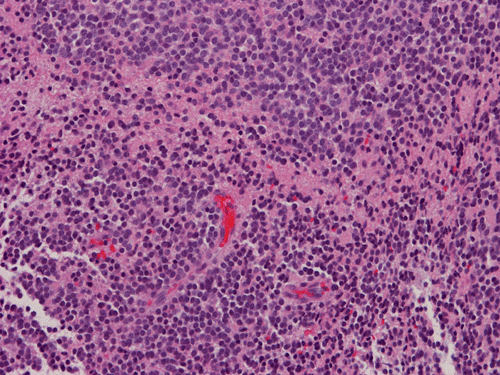
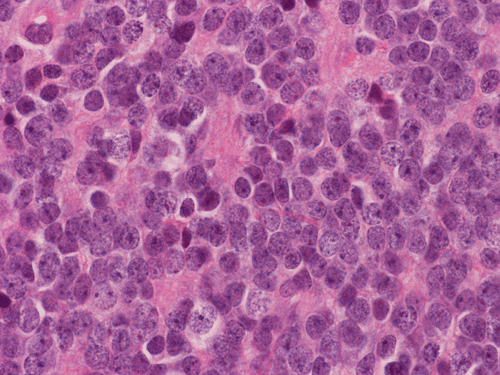
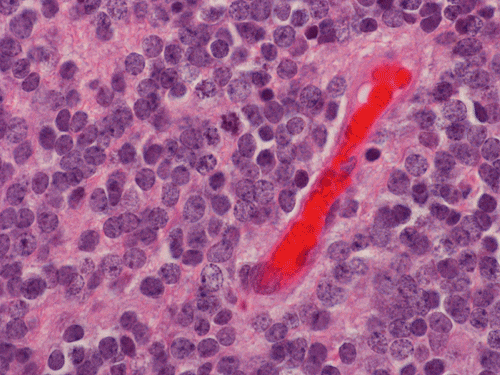
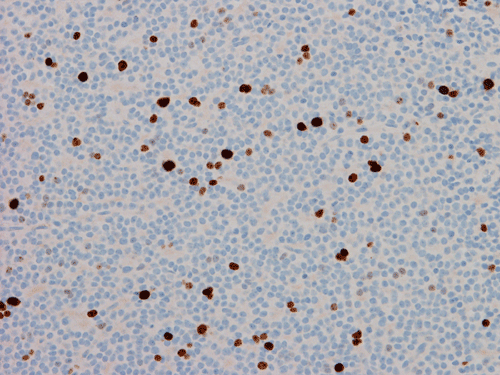
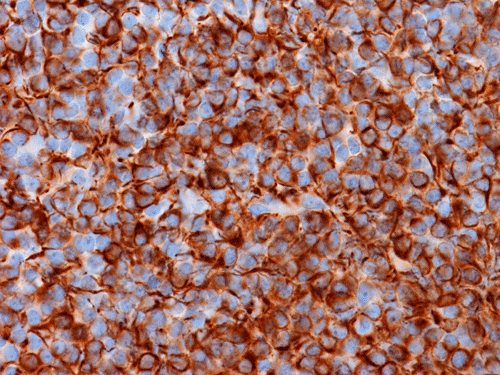
Pathology of the Case: The tumor is composed of neoplastic small blue cells on the cytologic preparation (Pancel C and D) with only minimal amount of cytoplasm. On frozen section (Panel E and F), the tumor cells are packed in solid sheets without specific pattern of arrangement. On permanent sections, the tumor again is composed of solid sheets of small blue cell tumors with rather monotonous nuclei (Panel G and H). In some areas, there is some small neuropil islands (Panel I). No distinct perivascular coronary arrangement is noted. (Panel J). There is a moderate labeling index on Ki67 (Panel K). The tumor cells are strongly positive for neurofilament proteins (Panel L). Some large stellate, cells positive for glial fibrillary acidic protein (GFAP) are also present and these cells may represent reactive astrocytes (Panel M).
| DIAGNOSIS: Pineal parenchymal tumor of intermediate differentiation, WHO grade II. |
Discussion:
General Information
Pineal tumors are relatively uncommon tumors and are variably estimated to constitute between 0.5 -3% of of all intracranial tumors. Pineal germinoma is perhaps the most common one. Its high incidence in children makes pineal tumors more commonly seen in children. Primary pineal tumors other than germinoma are even rarer. Pineoblastoma, pineal parenchymal tumor of intermediate differentiation (PPTID), papillary pineal tumor, and pineocytoma comprise a spectrum of tumor that spans from high grade malignancy to benign (WHO grade IV to grade I) 1. A spectrum of maturation is also demonstrated by these tumors. PPTID and papillary pineal tumor have variable biological potential and can be WHO grade II or III. In contrast, pineal cysts are quite common and often found as incidental findings, reported to be present in up to 40% of routine autopsies 2. Generally pineal cysts are asymptomatic, although they are occasionally known to bleed or to cause compression.
Currently PPTID is the largest subgroup of the pineal parenchymal tumors and comprises 20% of all pineal parenchymal tumors with a peak incidence in early adulthood and a slight female predilection 1. Clinical manifestations are those typical of pineal region tumors. Although PPTID is described as a "potentially aggressive neoplasm", cerebrospinal and extraneural metastases are uncommon. The five-year survival of patients with PPTID is broadly characterized as 39-74% with prognosis varying based on morphologic subtype and histologic grading 1. A 2002 study by Lutterbach et al. which reviewed 101 cases of PPTID and their treatment reported an overall median survival time of 100 months (or 8.3 years) with variability of survival time dependent on disease extent, tumor differentiation and residual disease post-treatment (treatment consisted of varying combinations of surgical resection, radiotherapy and chemotherapy). All three criteria were found to be independent risk factors for a poor prognosis 3.
Neurophysiological Correlation
The pineal gland is an endocrine gland located in the posterior extremity of the roof of the third ventricle. It secretes melatonin which is involved in regulation of the sleep/wake cycle and other behavioral cycles. Information about light entering the eyes is passed along the optic nerve to the midbrain, then to the suprachiasmic nucleus of the hypothalamus, the superior cervical ganglion and the pineal gland. Light input via this pathway suppresses the production of melatonin by the pineal gland and causes a wakeful state. Absence of light input via this pathway results in the production of melatonin by the pineal gland and triggers the sleep cycle 4.
Clinical Manifestations
Symptoms and signs are usually a reflection of compression and destruction of the pineal and the surrounding structures. Due to the strategic location clinical manifestations often points to hydrocephalus due to the obstruction of the aqueduct, compression of the upper brainstem and cerebellum. Clinical symptoms and signs include sleep disturbance, headaches, vomiting, diplopia and disorders of extraocular movement, including Parinaud Syndrome. If the tumor involves the adjacent hypothalamus, vision loss, visual field defects, growth failure, precocious puberty, diabetes insipidus and weight loss can also occur.
In addition, germ cell tumors frequently present with long prodromes of endocrine dysfunction and central diabetes before the neurological signs come into attention 5, 6. It should also note that patients with Klinefelter's syndrome are predisposed to cerebral germinomas, frequently in the pineal region 7.
Parinaud Syndrome
Parinaud syndrome, also known as dorsal midbrain syndrome, is name after Henri Parinaud (1844-1905), the famous French ophthalmologist. The etiology is disturbance of function of the upper brainstem around the pineal regions. It is most commonly associated with tumors of the midbrain and pineal region, multiple sclerosis and ischemic strokes of the upper brainstem. It can, however, also be associated with other causes of obstructive hydrocephalus, thalamic tumors, midbrain hemorrhage, arteriovenous malformation, brain trauma, giant aneurysms of the posterior fossa, toxoplasmosis of the brainstem, neurosyphilis and tuberculomas. Vertical supranuclear ophthalmoplegia has also been associated with metabolic disorders, including Wernicke’s encephalopathy, Niemann-Pick disease, Tay Sachs, maple syrup urine disease, Wilson's disease, kernicterus and barbiturate overdose.
The clinical features of Parinaud syndrome results from disruption of the function of the dorsal upper midbrain or tectum and is characterized by the triad of impaired upward gaze, loss of pupillary light reflex and weakness of convergence. Parinaud syndrome can include other clinical manifestations including preference for downward gaze, preservation of near reflex, convergence retraction nystagmus, impaired visual pursuit, lid retraction (Collier's sign), and bilateral papilledema. With continued tumor expansion, downgaze and near reflex can also become impaired. In rare cases of pineal tumors, downward gaze impairment may occur without impairment of upward gaze.
Neuroanatomically, the pineal gland is directly above the vertical gaze center and light reflex center. As pineal tumors expand in the direction of least resistance, they cause pressure on these centers, compressing the superior colliculus (where the nucleus of the oculomoter nerve (CN III)and Edinger Westphal nucleus are located), structures of the periaqueductal gray matter and associated fibers of the posterior commissure, medial longitudinal fasiculus and the interstitial nucleus of Cajal. Convergence retraction nystagmus is also caused by the compression of fiber tracts and the resulting disinhibition of the ocular motor nuclei and group firing of the external ocular muscles. As the medial rectus is the most powerful of the extraocular muscles, co-firing of occulomotor nerveswill cause convergence and retraction of the globes into the orbits. Having patients visually track stripes on an optokinetic drum can be used to test for convergence retraction nystagmus.
It should be noted that impairment of upward gaze can result from defects in either the voluntary or involuntary vertical gaze center. The voluntary vertical gaze center is located in Broadmann's Area 8 of the frontal lobe, while the involuntary vertical gaze center is located in the midbrain. The oculocephalic or Doll's head reflex can be utilized to differentiate deficits of voluntary vertical gaze centers from deficits of the involuntary vertical gaze center, at least initially. Eventually with increased tumor expansion all aspects of vertical gaze may be impaired.
Pathology
Pineoblastoma (WHO grade IV), PPTID (WHO grade II-III), papillary pineal tumor (WHO grade II-III), and pineocytoma (WHO grade I) comprise a spectrum of small blue cell tumors with variable level of differentiation and malignant potential.
In essence, PPTID is composed of diffuse sheets or large lobules of uniform cells with mild to moderate nuclear atypia and low to moderate levels of mitotic activity. Differentiation of grade II and III tumors can be made based upon a combination of mitotic activity levels and neurofilament protein immunoreactivity. Rare mixed tumors exhibiting characteristic elements of both pineocytoma and pineoblastoma have also been classified as PPTID, albeit somewhat controversially. The tumor classification of PPTID originated in 1993 with the work of Schild, et al. reviewing data on 30 patients with pineal parenchymal tumors diagnosed between 1939 and 1991. Of the 30 pineal parenchymal tumors, four were sub-classified as PPTID by Schild and his colleagues 9.
Differential Diagnosis
Tumors of the pineal region can generally be classified into three major groups: tumors of germ cell origin, tumors of pineal cell origin and tumors of other cell origin.
Tumors of germ cell origin are the most common type of pineal tumors, comprising 40-50% of all pineal tumors. They can be further sub-classified as germinomas and non-germinomas (a group encompassing mature teratomas, malignant teratomas, embryonal cell carcinomas, endodermal sinus tumors, choriocarcinomas and mixed germ cell tumors). A pure germinoma is the most common pineal neoplasm. Pineal germinomas typically occur within the first three decades of life, with an increased incidence in Japan (16%). In general, germinomas have a strong male predilection and pineal germinomas exhibit an over 90% male predominance 2 with suprasellar pineal germinomas being a curious exception and equally prevalent in both sexes. Pineal germinomas exhibit both a tendency to disseminate via the cerebral spinal fluid and invade nearby brain tissue, and yet they are highly radiosensitive with a high survival rates. Intratumoral hemmorrhage is common.
PPTID has a histopathologic picture of a small blue cell tumor. However, the degree of pleomorphism is not as high as pineoblastoma or medulloblastoma. Particularly for the one with better differentiation, the cells would have overall features suggestive of lymphocytes. This would further suggest germinomas as they are associated with a substantial amount of lymphocytes. Search for large germinoma cells would be the diagnostic clue to distinguish these two categories. Histological distinction of PPTID with teratomas, yolk sac tumors, embryonal cell carcinomas, and choriocarcioma components in germ cell tumors is relatively straightly forward. The table below provides comparison of different types of pineal parenchymal tumors.
Table: Histologic features of Pineocytoma, parenchymal parenchymal tumor of intermediate differentiation, papillary tumor of the pineal tumor, and pineoblastoma.
|
|
Pineocytoma |
PPPTID |
PTPR |
Pineoblastoma |
|
Incidence |
Relatively common among pineal parenchymal tumor.s |
Relatively common among pineal parenchymal tumors. |
Rare among pineal parenchymal tumors. |
Common among pineal parenchymal tumors. |
|
Age |
Any age but mainly seen in adult (mean age: 38 years) |
Any age but mainly seen in adult (mean age: 38 years) |
Most common in young adults in the 2nd and 3rd decades. |
Most common in infants, children, and young adults. |
|
WHO Grade |
I |
II-III |
II-III |
IV |
|
Prognosis |
5-year survival rate is about 86% to 100%. |
5-year survival rate is about 39% to 74%. |
5-year survival rate is about 72%. |
5-year survival rate is about 58%. |
|
Histologic features |
Moderately cellular, well-differentiated, small, monotonous, cells that resemble pineocytes. Low grade round nuclei. Moderate amount of cytoplasm. Conspicous cytoplasmic processes with club shaped-expansion that can be demonstrated by silver stain or immunohistochemistry for neurofilament. No or rare mitosis.
|
Moderately to highly cellular with mild to moderate nuclear atypia. Pineocytomatous pattern can be present. Tumor cells can have a neurocytoma like diffuse pattern or a lobulated pattern. Mitotic figures are present and are more common in grade III than grade II tumors. |
An epithelial appearing tumor with papillary architecture which may closely simulate ependymoma. Pleomrophic nuclei may be present. Mitotic figures range from 0-10/10 HPF. Necrotic areas are often seen. Hyalinized vessels are common but endothelial proliferation is not seen. |
Basically a high grade small blue cell tumor with minimal amount of cytoplasm, morphologically similar to medulloblastoma. Homer Wright and Flexner-Wintersteiner rossete can be seen. Mitoses are usually common. Necrosis is common. Melanin production, cartilaginous, and rhabdomyoblastic differentiation can be encounter. These tumors are often referred to as “pineal analage tumors”. |
|
Pineocytomatous Rosette |
Yes |
No |
No |
No |
|
Immunohistochemistry |
Positive for synaptophysin, neurofilament, and specific photoreceptor antigens. |
Positive for synaptophysin, neurofilament, and photoreceptor specific antigens. Grade II tumors tend to be positive for neurofilament but not grade III tumors.
|
Synaptophysin and chromogranin are sometimes weakly positive, neurofilament is negative. In contrast to ependymoma, PTPT is positive for cytokeratin (AE1/AE3, Cam5.2, CK18) particularly in papillary structures; in addition, only focal positivity for GFAP would be seen. Also positive for S-100, MAP2, N-CAM, and transthyretin. Focal membrane or dot like positivity for EMA can sometimes be seen. |
Positive for synaptophysin, neurofilament, and specific photoreceptor antigens. |
*This table is based on Reference 1.
Tumors of pineal cell origin include the pineocytoma, the pineoblastoma, the parenchymal pineal tumor of intermediate differentiation and papillary tumors of the pineal region. Individual parenchymal tumors are less common than germinomas or teratomas. These tumors originate in the neuroepithelial cells of the pineal gland and can exist within the same tumor and in transitional forms. They can also contain melanin. Pineocytomas (WHO grade I) are rare, slowly growing neoplasms consisting of small, uniform, mature-appearing pineocytes and occurring mainly in adults. These tumors are generally well-defined, non-infiltrative lesions. They are solid lesions, less cellular than pineoblastomas, with greater amounts of cellular cytoplasm. When the tumors exhibit greater nueronal differention they are more benign.
Pineoblastomas (WHO grade IV) are highly malignant primitive embryonal tumors frequently associated with CSF dissemination and composed of dense, patternless sheets of small, poorly differentiated, immature cells with slightly irregular nuclei and scant cytoplasm. Leptomeningeal and subependymal seeding is often present at the time of diagnosis 2. Pineoblastomas occur mainly in children and often exhibit focal hemorrhage and microscopic necrosis. Papillary tumors of the pineal region (WHO grade II-III) are rare neuroepithelial tumors of the pineal region typically occurring in adults and possessing a papillary architecture, an epithelial cytology and an immunopositivity for cytokeratin.
Tumors of other cellular origins arising in the pineal region include gliomas (generally astrocytomas), meningiomas and metastatic tumors. Pineal region glial tumors and meningiomas usually extend from the adjacent brainstem or tentorium and only rarely originate from the pineal gland itself 2. Metastatic tumors to the pineal region are relatively rare. The trilateral retinoblastoma is one example of a highly malignant and aggressive tumor which involves the retinas of both eyes (bilateral retinoblastomas) as well as the pineal gland (pineoblastoma); the pineal region mass is considered by some to be metastatic.
The MRI is the diagnostic study of choice for detecting pineal region tumors. Pineal region masses display highly characteristic imaging which is both indicative of their pathology and suggestive of their specific type. These imaging characteristics are presented in Table A. (Atlas, 2002, p 662-663). Pineal germinomas tend to be well-cicumscribed relatively homogenous lesions inseparable from the pineal body itself. They are typically low intensity (isointense to gray matter) on T2-weighted images and may exhibit intratumoral hemorrhage and invasion of the adjacent brain with associated edema and subarachnoid dissemination. Post-IV contrast, germinomas and their metastases typically enhance markedly and homogenously. Pineal germinomas do not calcify or contain cysts; although calcification of the pineal body itself is not uncommon. (Atlas, 2002, p664)
MRI is useful in distinguising pineal neoplasms from pineal cysts as pineal neoplasms are depicted as lobulated solid tumors which enhance densely with contracts. Generally pineoblastomas are isointense to gray matter on T2 spin images. Pineocytomas (with their higher degree of cytoplasm) should have a relatively higher signal intensity on T2 weighted images. Intratumoral calcivaications can exist in both pineoblastomas and pineocytomas, but are more common in pineocytomas 2. On MRI, the pineal cyst is round with smooth margins; its contents are homogenous and either isointense to CSF or diffusely hyperintense.
Blood and CSF samples may reveal elevated Human chorionic gonadotropin (HCG) and alpha-fetoprotein (AFP) associated with tumors of germ cell origin and some metastatic tumors. Tumors of pineal cell origin are not typically associated with elevated biological tumor markers. In all cases of pineal tumors, tissue biopsy is required for definitive diagnosis.
Reference:
Nakazato Y, Jouvet A, BW Scheithauer. Tumors of the Pineal Region in . WHO Classification of Tumours of the Central Nervous System, Fourth Edition. edited by Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Lyon 2007.
Atlas, SW. Magnetic Resonance Imaging of the Brain and Spine. Volume I. 3rd Edition. Philadelphia: Lippincott Williams & Wilkins, 2002, pp. 662-671.
Lutterbach J, Fauchon F, Schild SE, Chang SM, Pagenstecher A, Volk B, Ostertag C, Momm F, Jouvet A. Malignant Pineal Parenchymal Tumors in Adult Patients: Patterns of Care and Prognostic Factors. Neurosurgery July 2002: 51: 44-56.
Allgood D. Pineal Tumors and their Clinical Manifestations. Clinical and Refractive Optometry. 2005 16:2.
Diniz, JS, Oliveira EA, Servilha MM. Diabetes insipidus as an early clinical manifestation of pineal tumor. Journal of Pediatrics 2000 76:383-6.
Tarng DC, Huang TP. Diabetes Insipidus as an Early Sign of Pineal Tumor. American Journal of Nephrology 1995; 15:161-164.
Arens, R , Marcus D, Engleberg S, Findler G, Goodman RM, Passwell JH. Cerebral germinomas and Klinefelter syndrome: A review. Cancer, 198815; 61:1228-31.
Schild SE, Scheithauer BW, Schomberg PJ, Hook CC, Kelly PJ, Frick L, Robinow JS, Buskirk SJ. Pineal parenchymal tumors. Clinical, pathologic, and therapeutic aspects. Cancer. 1993 72:870-80.