DiffQuick
DiffQuick
DiffQuick
DiffQuick
PAP Stain
PAP Stain
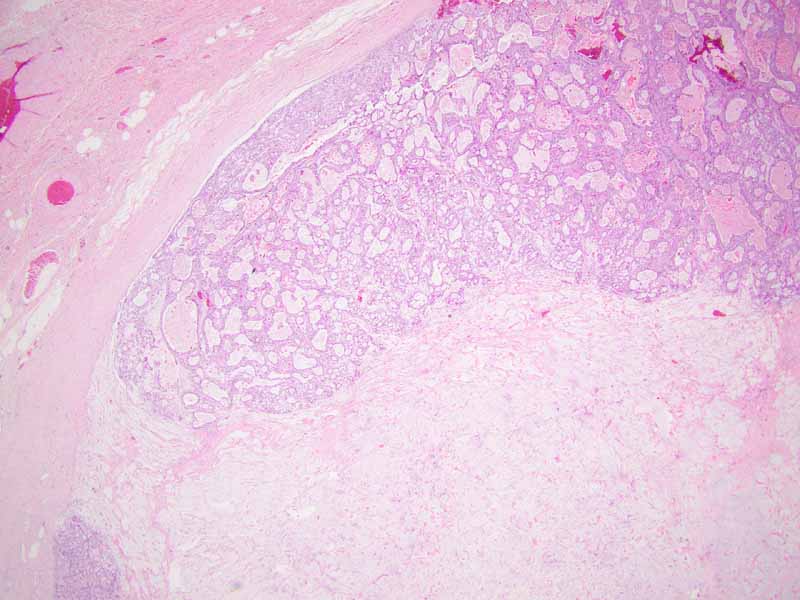
Cell block
TLE-1
| A 37 year-old Woman with an 8 cm Popliteal Mass. June, 2020, Case 2006-1. Home Page |
Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Clinical information: The patient was a 37 year-old woman with a deep 5.5 cm popliteal mass. An ultrasound guided FNA was performed and yielded the followings.
|
|
|
|
|
|
|
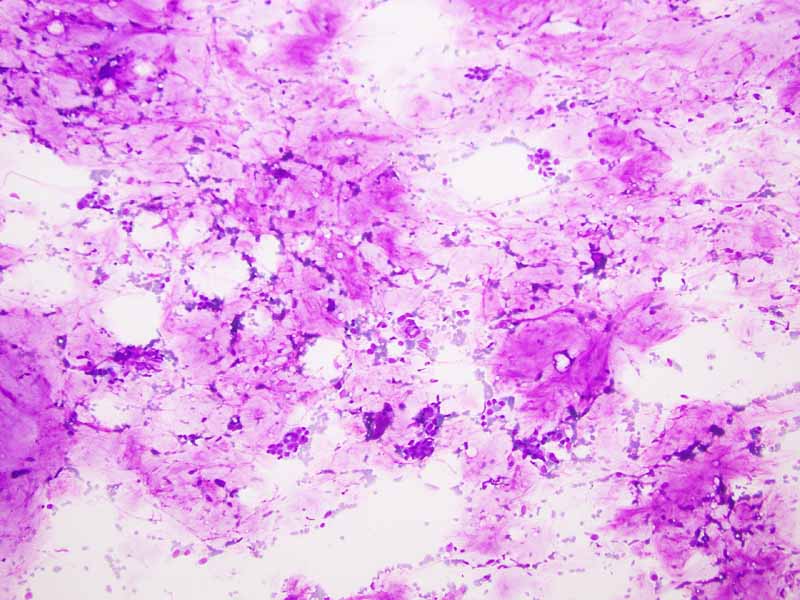
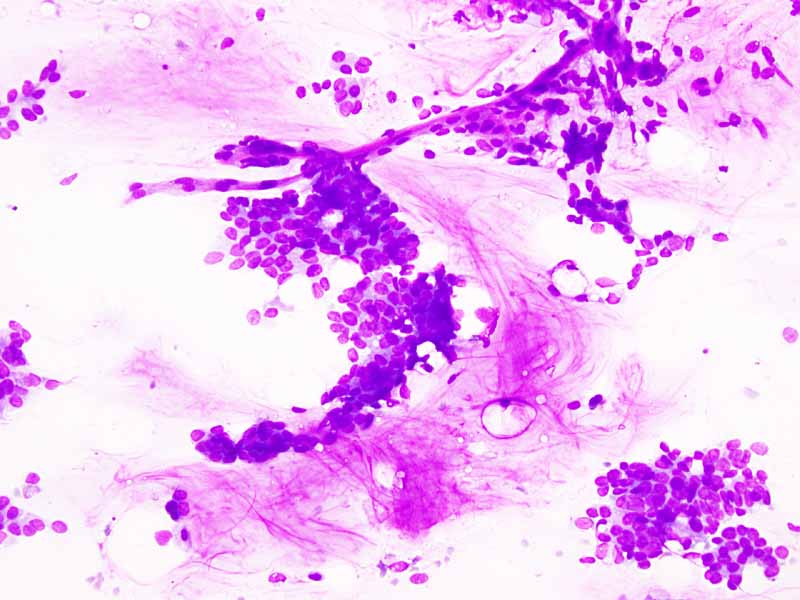
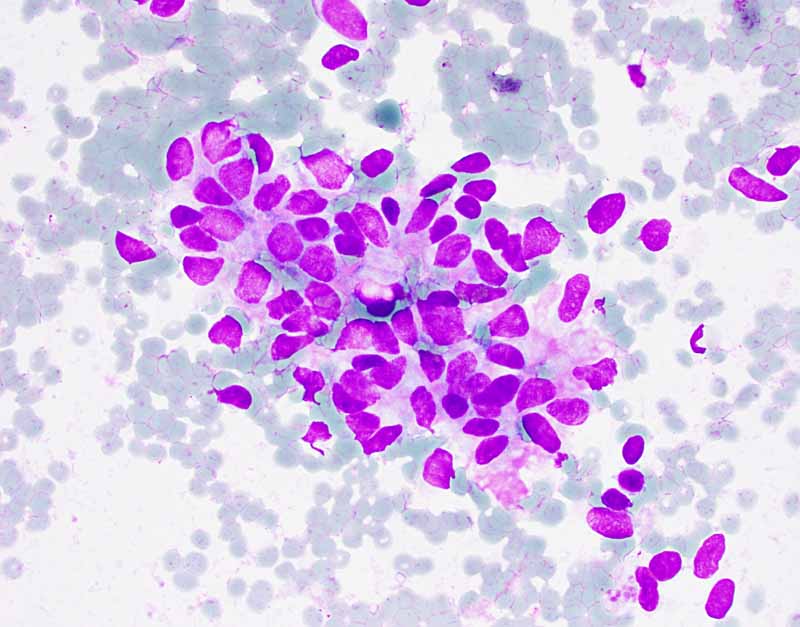
A. DiffQuick |
B. DiffQuick |
C. DiffQuick |
D. DiffQuick |
|
|
|
|
|
|
|
|
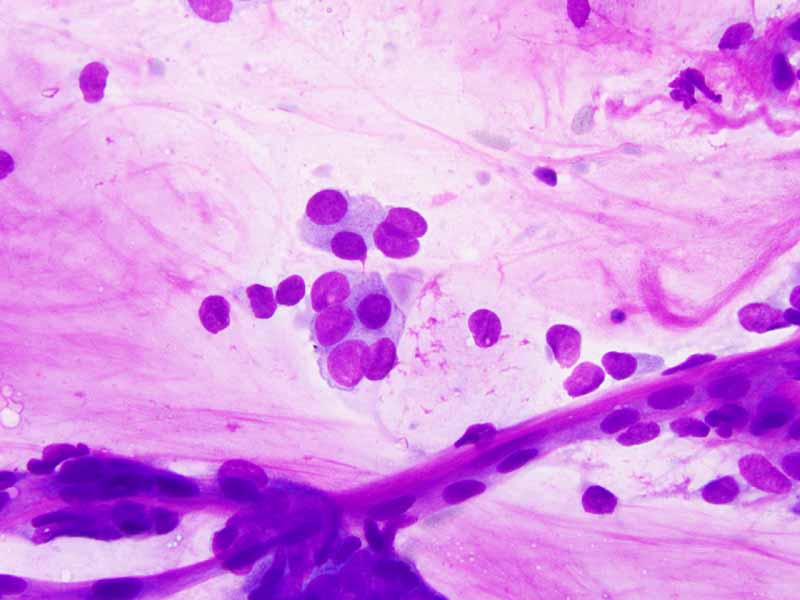
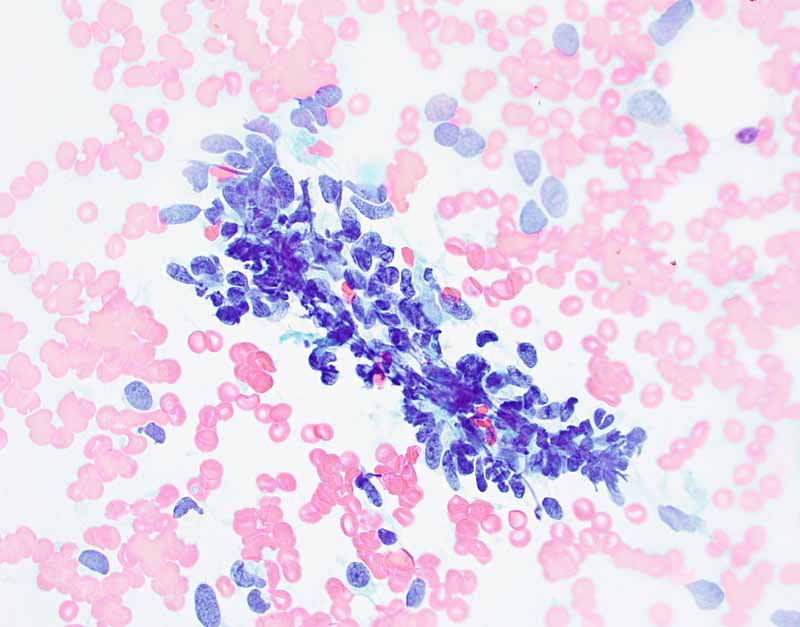
E. PAP Stain |
F. PAP Stain |
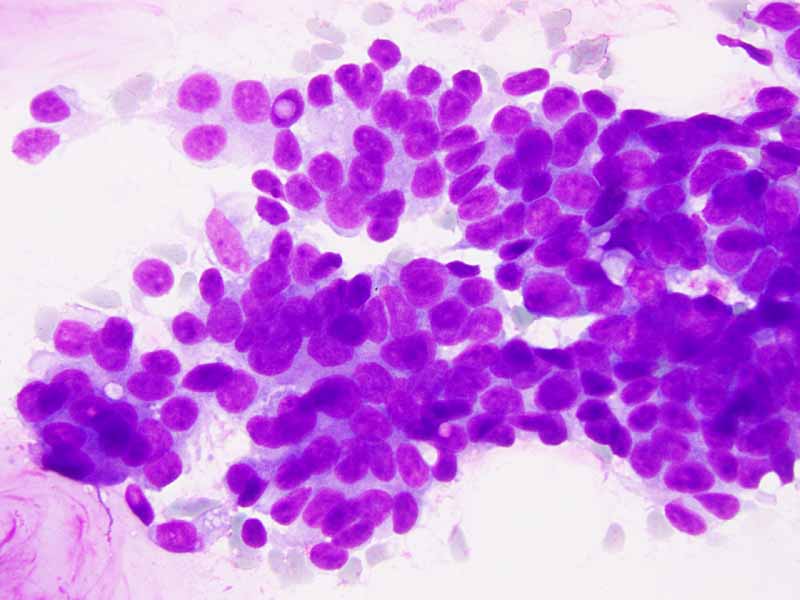
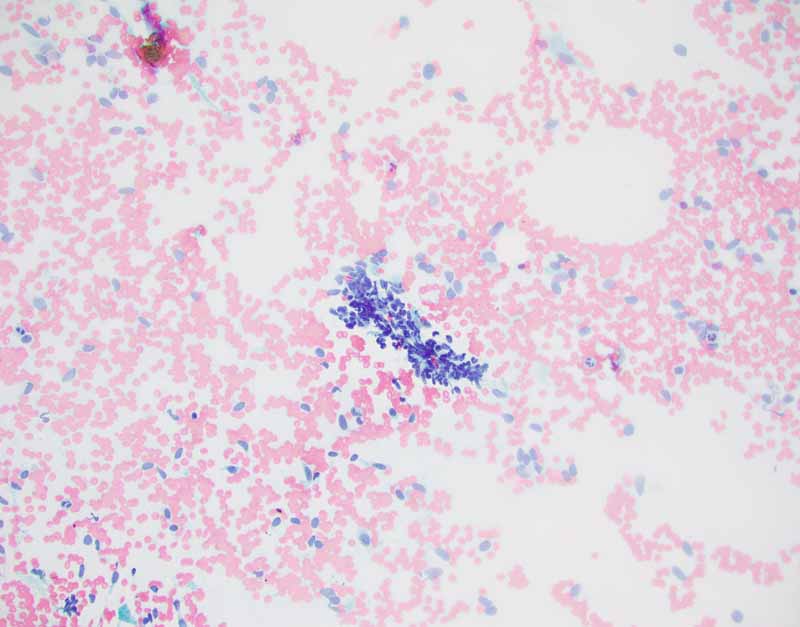
G. Cell block |
H. TLE-1 |
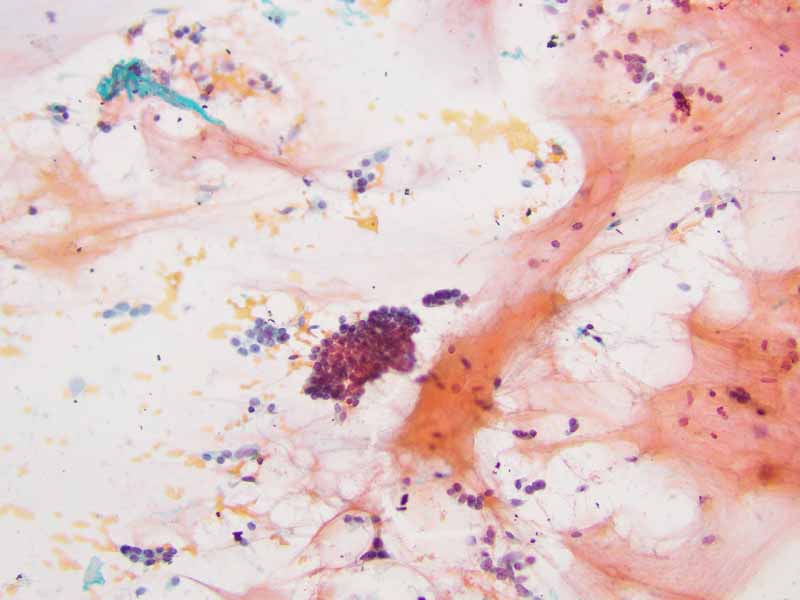
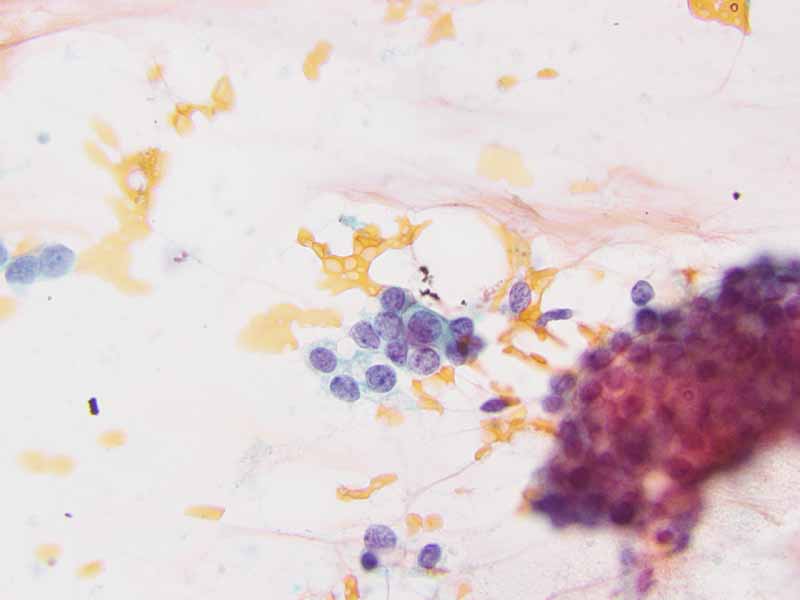
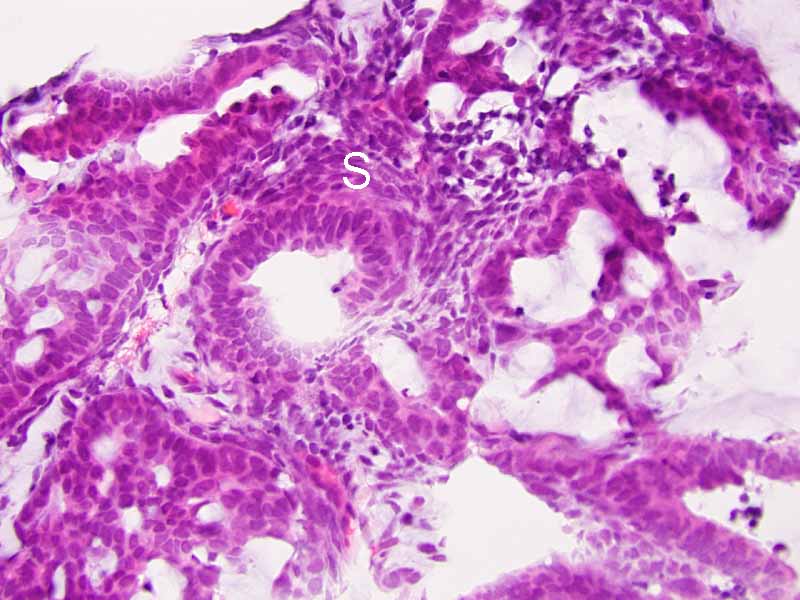
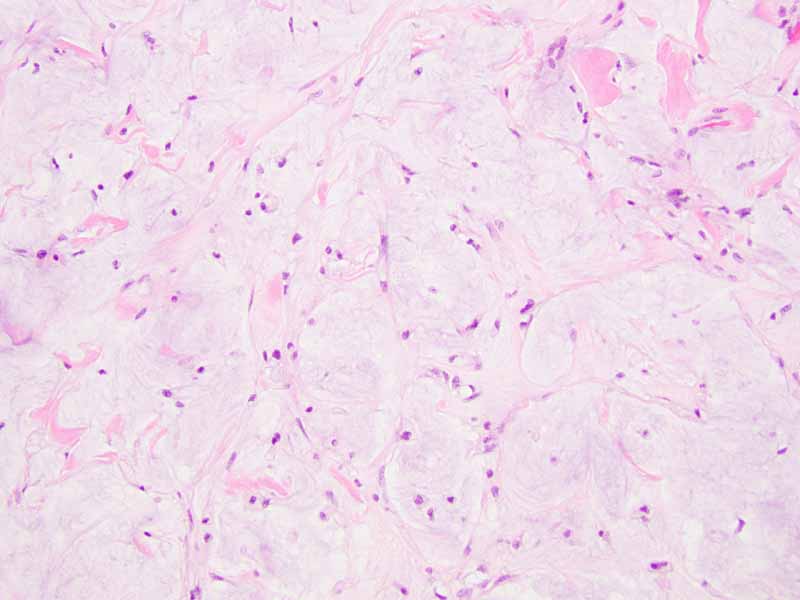
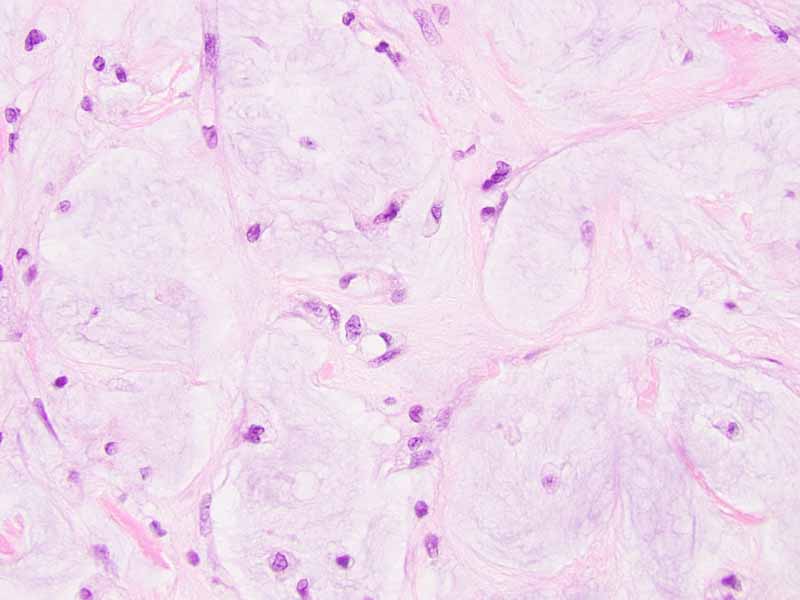
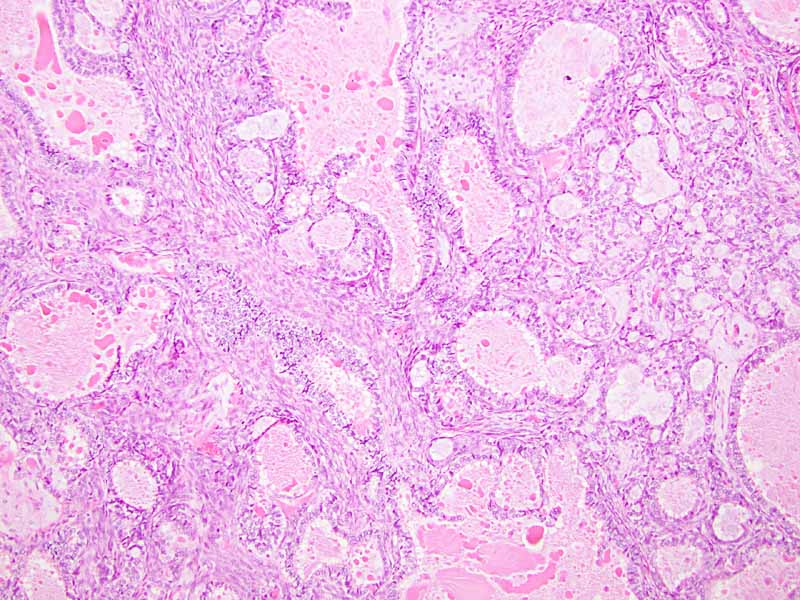
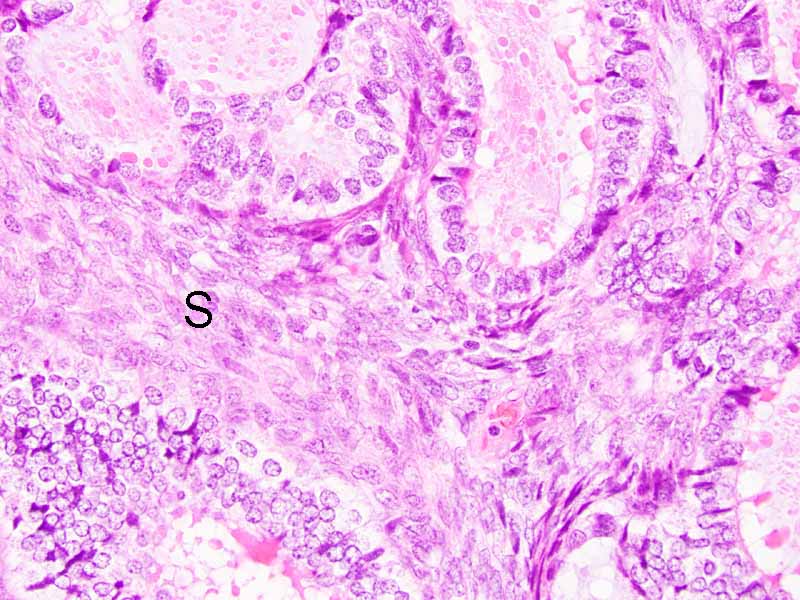
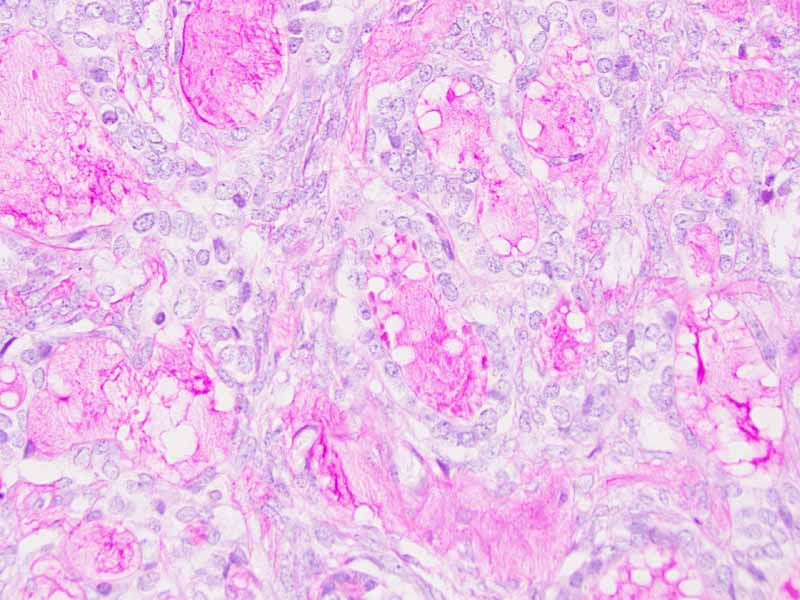
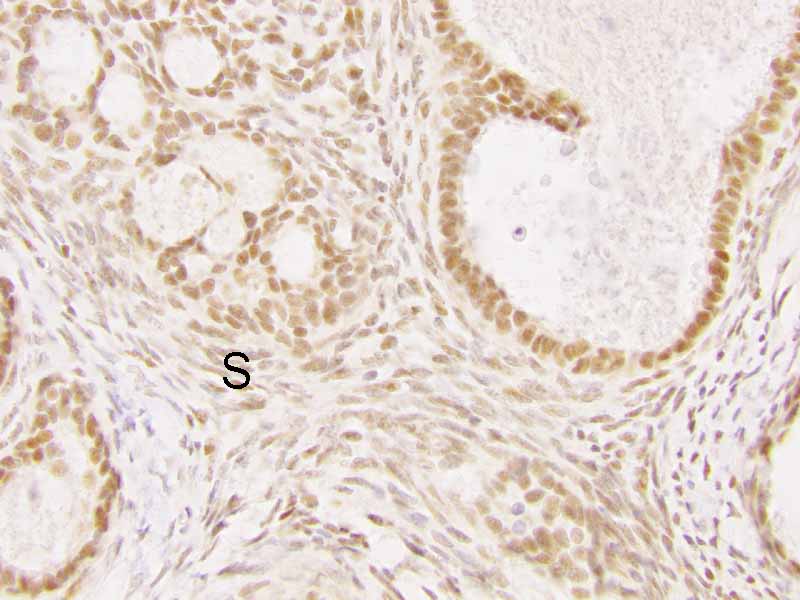
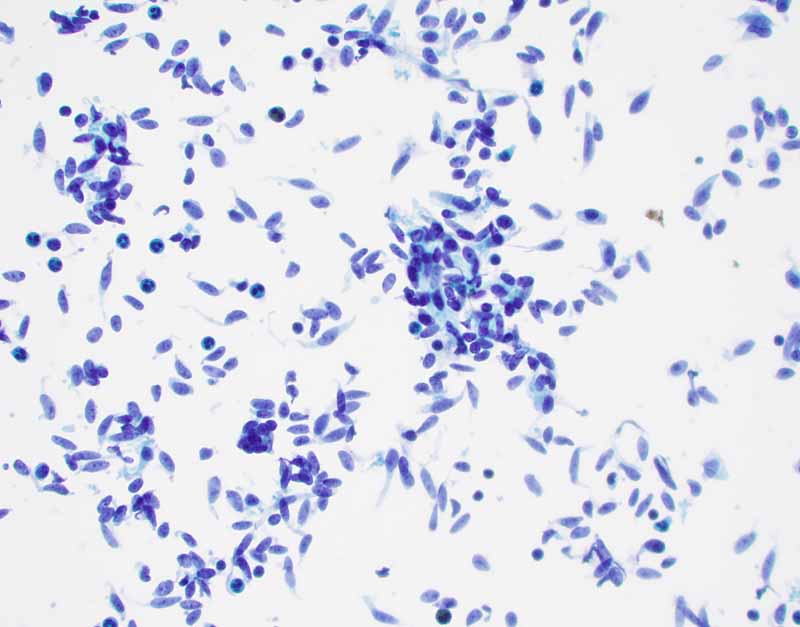
Cytology of the Case: The background is dominated by a substantial amount of mucoid material (Panel A) and admixed with these materials are small clusters of neoplastic cells. Some delicate blood vessels are also present (Panel B). A small number of tumor cells are adhered to these blood vessels but there is a lack of genuine papillary arrangement. The cells are epithelioid and contains a moderate amount of cytoplasm (Panel B, C, D, E, and F) that are finely vacuolated (Panel D and F) in some of them. The neoplastic cells are rather adhesive to each other. The nuclei are hyperchromatic and without pseudonuclear inclusion in most of them. There is no prominent nucleoli A minute amount of tissue is present in the cell block (Panel G). There is definitive gland formation. Some spindle neoplastic cells (S in Panel G) are also present next to the glandular structure. The nuclear features of these cells are identical to that of the glandular cells.
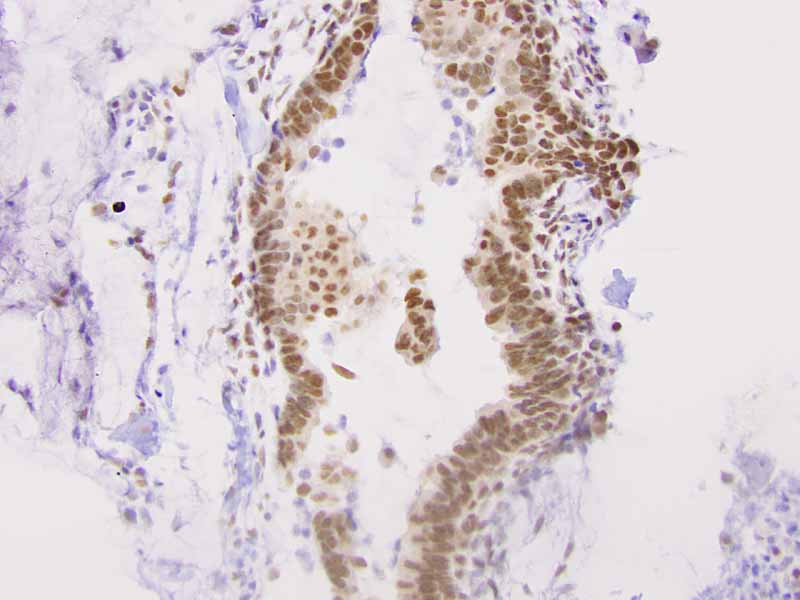
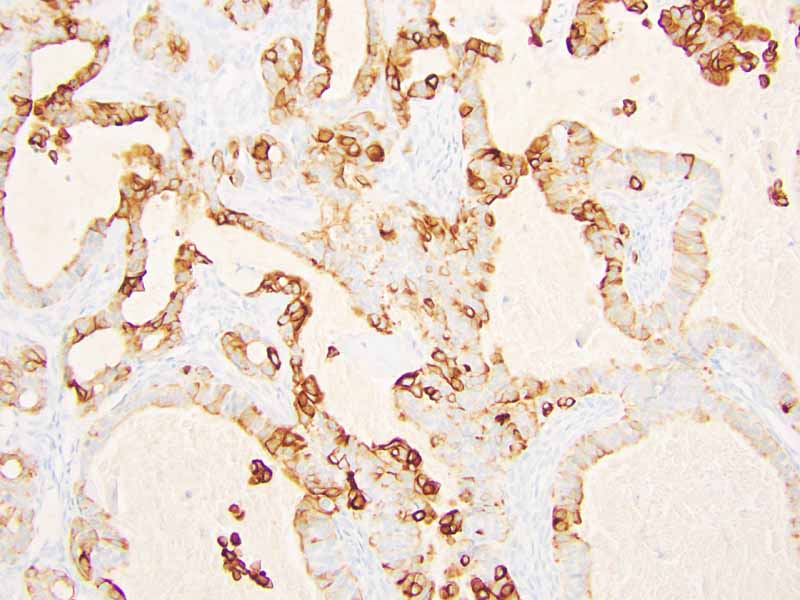
Immunohistochemistry: The tumor cells are positivefor TLE-1 (nuclear staining) (Panel H) and cytokeratin 7 but negative for GCDFP-1.
Cytogenetics: FISH was performed and no rearrangement of SS18 was demonstrated.
 |
 |
 |
 |
 |
 |
|
I. Online slide |
J. | K | L. | M. | N. |
 |
 |
 |
 |
 |
|
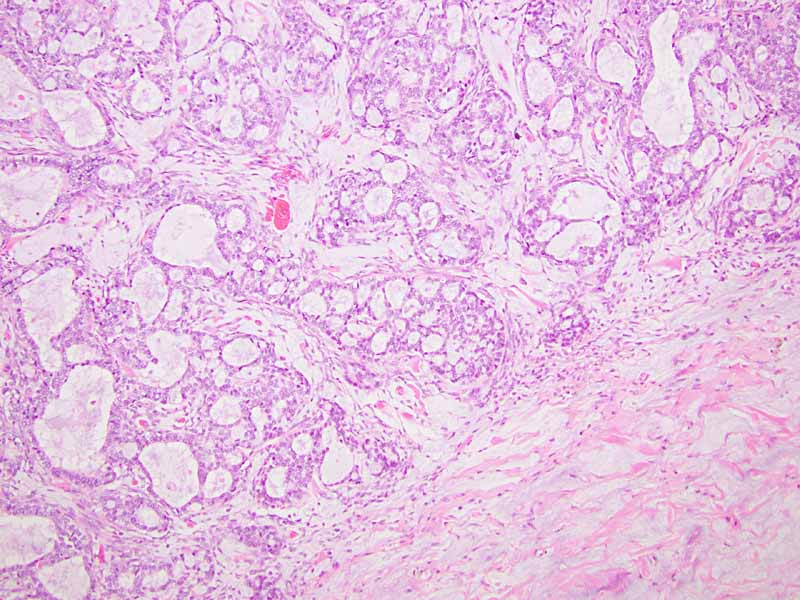
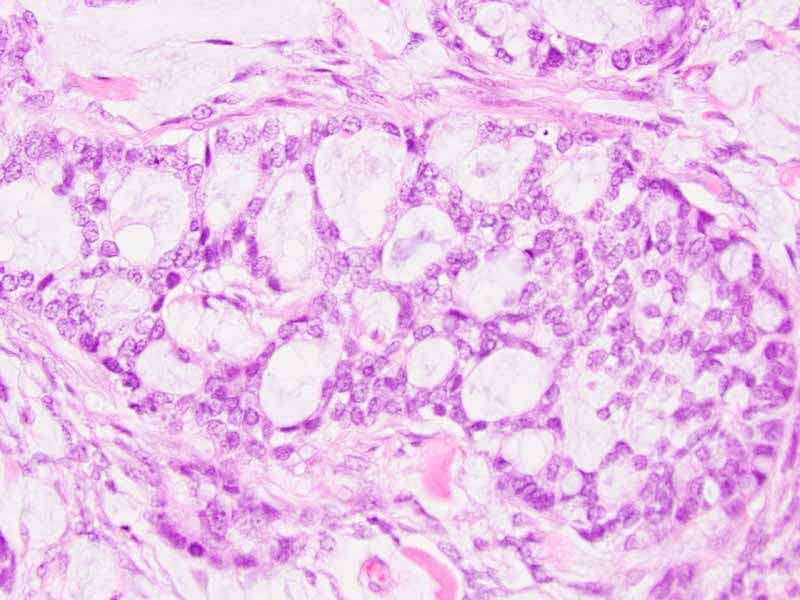
| O. | P. |
Q. PAS |
R. Cytokeratin 7 |
S. TLE-1 |
 |
 |
 |
 |
|
|
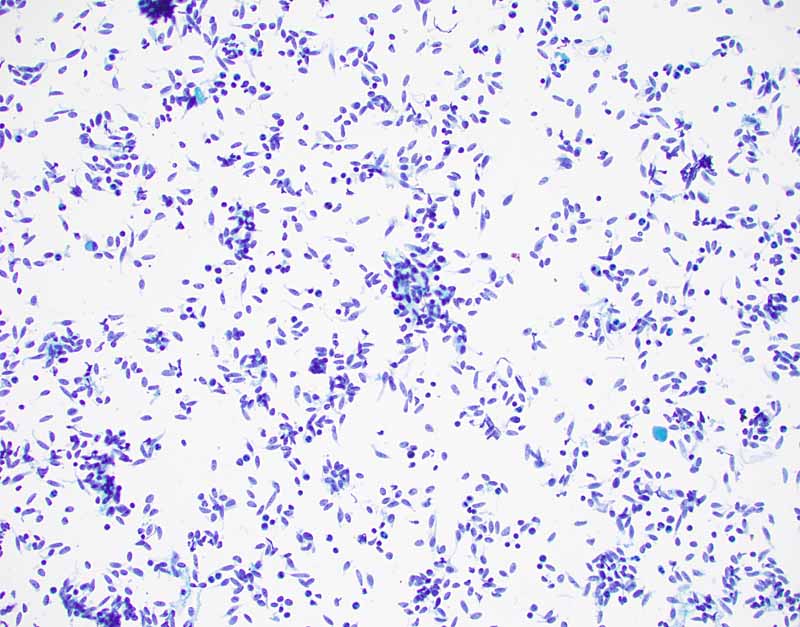
1. DiffQuick |
2. DiffQuick |
3. PAP Stain |
4. PAP Stain |
|
 |
 |
 |
 |
|
|
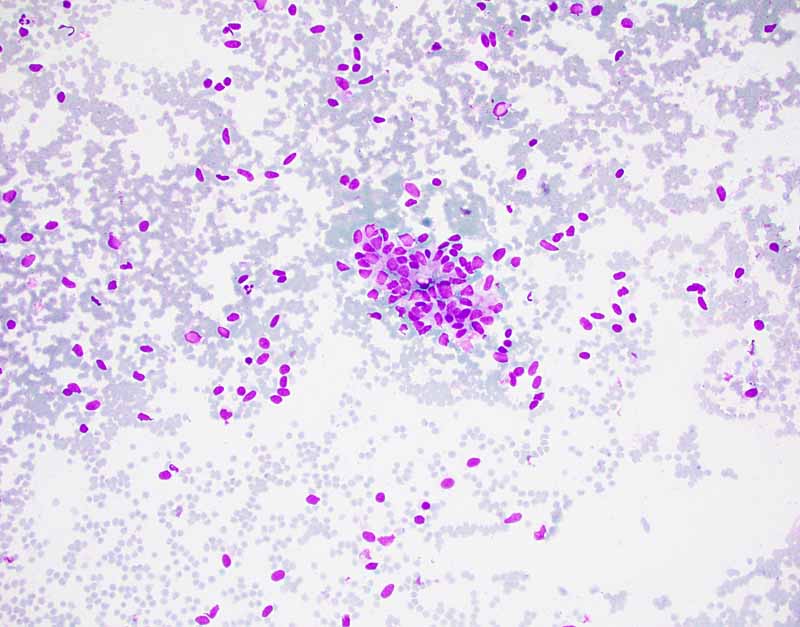
5. Cytospin |
6. Cytospin |
7. | 8. |
| DIAGNOSIS: Biphasic synovial sarcoma. |
Discussion:
General Information
Pathology
Immunohistochemistry
Molecular Pathology
Differential
diagnosis
Related Interesting Cases
Synovial sarcomas
[Review:
Thway K and Fisher C, 2014] is an uncommon but not rare tumor. This
entity spans a spectrum of histopathologic features and immunohistochemical
profile. With its propensity to occur in almost any site, correct diagnosis can
be a diagnostic challenge. It is relatively sensitive to chemotherapy in
comparison with other sarcomas so a correct diagnosis is important.
Synovial sarcoma
are most commonly seen in younger patients with slight male predominance. It
typically occurs in para-articular region of large joints particularly the knee
but it occurs in almost any site. However, it is neither common in joint
cavities nor in arears with apparent close association with synovial tissue.
Although it is one of the first sarcomas to be defined by the presence of a
specific chromosomal translocation involving
SS18(SYT) typically SS18-SSX1 and SS18-SSX2, these genetic changes
are not detected in 100% of synovial sarcomas.
Synovial sarcomas span the spectrum from monophasic fibrous type to biphasic
type with both distinct epithelial and spindle cell components in varying
proportion, to epithelial-predominant type to poorly differentiated (round cell)
type. Both the epithelial-predominant type and poorly differentiated type are
uncommon. Monophasic fibrous type is the most common.
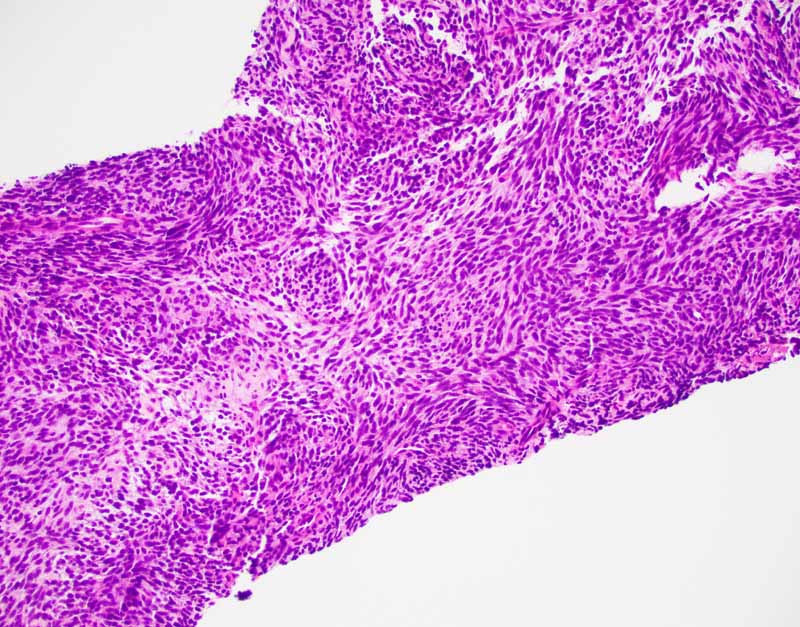
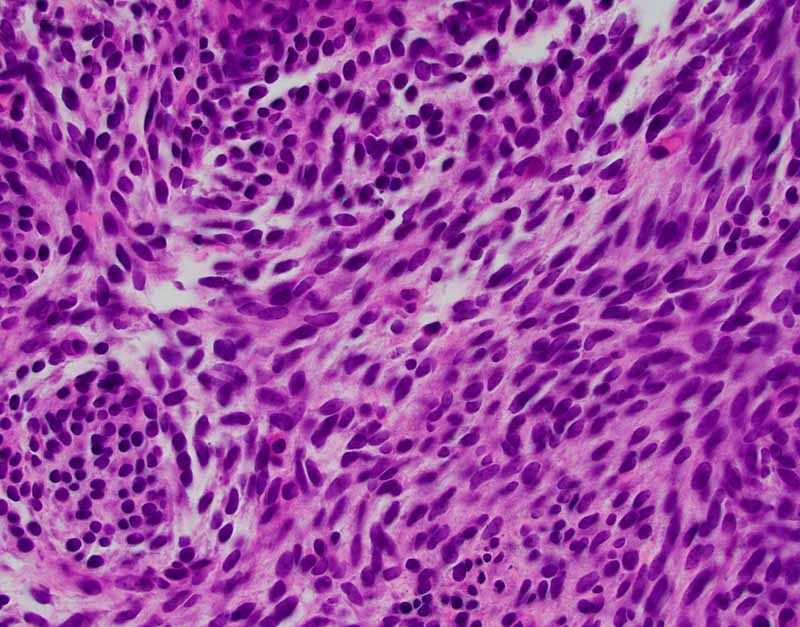
The classic biphasic type is readily recognizable by the presence of
epithelial component in a background of spindle cells similar to fibroblasts.
Well-defined sharp transition rather than gradual transition is typical between
the epithelial and spindle cell component. The glandular growth pattern can vary
from obvious to vague and, in the later case, clefts resulted from shrinkage due
to processing must not be mistaken as genuine glandular lumens. The glandular
epithelial cells are usually cuboidal to columnar. Secretions within the lumen
can be seen and squamous metaplasia may be present. The spindle cell component
is typically composed of monotonous well-oriented plump spindle cells with
indistinct cytoplasm and oval to elongated hyperchromatic nuclei. Fibrotic areas
with hyalinization and myxoid changes can be found. In about 20% of cases,
calcifications with or without ossification is present and act as a good
diagnostic clue. Substantial number of mast cell is another diagnostic clue.
Monophasic fibrous synovial sarcoma is typically composed of monotonous solid
sheets of small spindle cells with indistinct cytoplasmic membrane and small
hyperchromatic nuclei without nucleoli. Vague palisading arrangement can be
present. Usually, there is no intervening collagen fibers. Some foci of
epithelioid morphology can be seen. Secondary changes such as calcifications and
myxoid changes as seen in the spindle cell component of the biphasic type can
also be found. Staghorn shaped blood vessels similar to those in
hemangiopericytoma can be part of the histopathologic picture.
Epithelial-predominantly synovial sarcoma, also known as monophasic epithelial
synovial sarcoma, is a rare entity. They closely mimic carcinoma and can be a
diagnostic pitfall. This type of synovial sarcoma typically contains a small
amount of spindle cell component that worth to be hunted down. The term
monophasic epithelial synovial sarcoma may well be theoretical concept.
Poorly
differentiated areas can superimpose on any of the three subtype to constitute a
poorly differentiated synovial sarcoma. This entity spans a spectrum of
histologic features from small cell to spindle cell, to large cell or
epithelioid with round nuclei and prominent nucleoli. Rhabdoid features can be
part of the histologic feature. It is, in reality, a high-grade transformation
of synovial sarcomas. In one study, about 62% of synovial sarcoma had poorly
differentiated areas and the poorly differentiated component could represent up
to 90% of the tumors
[Machan
SK et al., 1999]. These tumors are more aggressive and have a higher
metastatic rate. When the high-grade component dominates the histopathologic
picture, the underlying synovial sarcoma can be difficult to be recognized.
TLE1 is a highly
sensitive but not entirely specific marker for the diagnosis of synovial sarcoma
[Foo WC et al., 2011;
Jagdis A et al., 2009;
Terry J et al., 2007] as TLE1 is expressed in other soft tissue and bone
tumors
[Kosemehmetoglu K et al., 2009] including endometrial stromal sarcoma, acral
myxoinflammatory fibroblastic tumor, schwannoma, liposarcoma, and others. CD99
is positive in about 60%-70% of synovial sarcomas
[Pelmus et al., 2002;
Olsen SH et al., 2006]. This feature can result in confusion with Ewing
sarcoma. Expression of either TLE1 or
CD99 has been demonstrated in some carcinomas but co-expression of these two
markers in carcinomas is
rare
[Zaccarini
DJ et al., 2018].
EMA may well be the most
sensitive marker for recognition of synovial sarcoma
[Pelmus et al., 2002;
Olsen SH et al., 2006]. Keratin and EMA are widely expressed by synovial
sarcomas
[Guillou L et al., 1997]. Expression is usually patchy rather than diffuse
but epithelial components are strongly positive. The number of positive cells in
monophasic fibrous synovial sarcoma can be scant and may requires extensive
sampling before being demonstrated. These markers are less expressed in poorly
differentiated synovial sarcomas
[Van de Rijn et at;, 1999]. Because of positive immunoreactivity for S100 in
some synovialsarcoma particularly the monophasic fibrous type, misdiagnosis as a
malignant peripheral nerve sheath tumor can be a diagnostic pitfall. Reduced
expression of SMARCB1/INI1 can be seen in synovial sarcoma regardless of whether
rhabdoid changes are present
[Kohashi K et al., 2010]. Synovial sarcomas are also positive for Bcl2
[Suster S et al., 1998] and calponin
[Fisher C et al., 2003].
Translocation involving
t(X;18)(p11.2;q11.2) translocation involving
SS18(SYT)
on chromosome 18 is present in over
90% of synovial sarcoma regardless of the type
[Sandberg AA et al., 2002]. SS18-SSX1
is more common than SS18-SSX2.
SS18-SSX2 is typically seen in
monophasic fibrous synovial sarcoma and
SS18-SSX1 is typically associated with biphasic synovial sarcoma
[Antonescu CR et al., 2000;
Kawai A et al., 1998;
Ladanyi M et al., 2002]. Both translocations can be reliably detected by
FISH or RT-PCR
[Amary MFC et al., 2007]. TLE1
is upregulated in synovial sarcoma
[Nielsen TO et al., 2002;
Nagayama S et al., 2002] and TLE1 can be detected by immunohistochemistry.
Rare synovial sarcoma with
typical histopathologic features and immunohistochemical profile but without
SS18-SSX fusion could be harboring
fusion with unusual transcripts that cannot be detected using routine methods. A
large study involving 243 patients association of
SS18-SSX2 with better overall survival
in patients with localized disease at the time of diagnosis then those with
SS18-SSX1
[Ladanyi M et al., 2002]. In another study,
SS18-SSX1 fusion was associated with
early, but not late recurrence [Canter RJ et al., 2008].
Although synovial sarcoma shows a spectrum of histopathologic patterns that
can result in diagnostic difficulties, immunohistochemistry for TLE-1 and FISH
or PCR for the detection of SS18 are two powerful tool to confirm the diagnosis.
The key to correct diagnosis is, therefore, a high index of suspicion. Given the
known success of chemotherapy on synovial sarcoma, correct diagnosis has
clinical importance.
Monophasic fibrous synovial sarcoma may resemble a number of spindle cell
neoplasms including sarcomatoid carcinoma, malignant peripheral nerve sheath
tumor (MPNST), leiomyosarcoma, hemangiopericytoma/solitary fibrous tumor, and
fibrosarcoma.
Sarcomatoid carcinomas usually have a much higher nuclear grade than
monophasic fibrous synovial sarcoma. Their nuclear grade is more comparable to
poorly differentiated synovial sarcoma. In contrast, expression of cytokeratin
in sarcomatoid carcinomas is usually minimal or absence and monophasic fibrous
synovial sarcomas typically have patchy to minimal expression of cytokeratin.
History and location of the tumor would also provide good clues for the
differentiation.
MPNST overlap with
monophasic fibrous synovial sarcoma in both histologic features and
immunohistochemistry. In particular, MPNST is variably positive for TLE1 and
broad spectrum cytokeratin. This feature may suggest monophasic synovial
sarcoma. On the other hand, S100 is expressed in some monophasic fibrous
synovial sarcoma. This creates another confusion. While loss of H3K27Me3 is
present in up to about 60% of MPNST, it can be seen in rare cases of monophasic
fibrous synovial sarcoma
[Pekmezci
M et al., 2017;
Schaefer IM et al., 2016]. When there is a doubt,
FISH or PCR should be employed to confirm the diagnosis.
The diagnosis of biphasic synovial sarcomas arising from classic locations
are usually not a diagnostic challenge. In uncommon sites, however, it can be
challenging. The differential diagnoses include carcinomsarcoma, and glandular
MPNST. In particular, when a biphasic synovial sarcoma occuring in peritoneum
and pleuropulmonary region, these biphasic tumors can mimic malignant
mesothelioma. Malignant mesothelialoma occurs in older patients and,
histologically, the glandular and spindle cell component usually display a
gradual rather than abrupt transition between the two components. WT1 is often
positive in mesothelioma but negative in synovial sarcoma.
When a biphasic synovial sarcoma is dominated by epithelial or glandular
component, metastatic carcinoma and adnexal tumor of skin must be seriously
entertained. Astute identification of the spindle cell component in between the
epithelial component and correlation with clinical and imaging features form the
key for correct diagnosis.
When classic synovial
sarcoma areas are identified in a poorly differentiated synovial sarcoma, the
diagnosis is less challenging but it is not uncommon for the poorly
differentiated component to make up over 90% of the tumor
[Machan
SK et al., 1999]. Poorly differentiated
synovial sarcoma mimic other high-grade tumors with similar features. Poorly
differentiated synovial sarcoma tend to mimic small round cell tumors such as
Ewing sarcoma, neuroblastoma, mesenchymal chondrosarcoma, alveolar
rhabdomyosarcoma, and high-grade, non-Hodgkin lymphoma. When large epithelioid
cells or rhabdoid cells are present in a poorly differentiated synovial sarcoma,
they may be confused with epithelioid sarcoma and extrarenal rhabdoid tumor in
addition to metastatic carcinoma and sarcomatoid carcinoma.
While intraosseous location
is common in Ewing sarcoma, it is rare in synovial sarcoma. Ewing sarcoma,
however, overlaps with poorly differentiated synovial sarcoma in terms of age of
onset, histologic features, and immunohistochemical profile. In particular, CD99
is positive in 60%-70% of synovial sarcomas (both cytoplasmic and membranous
staining)
[Pelmus
et al., 2002;
Olsen SH et al., 2006] and Ewing sarcomas also
express cytokeratin. However, aberration of EWSR1 is specific for Ewing sarcoma
and aberration of SS18 is specific for synovial sarcoma. These genetic tests
should be able to distinguish most of the cases.
Related Interesting Cases: