




ATPase pH 9.6
Trichrome
NADH-TR
SDH
COX


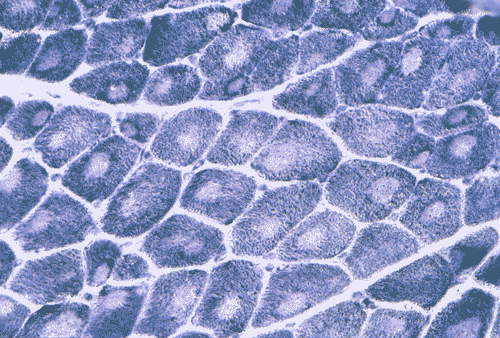
PAS
Desmin
Electron miroscopy
| A 5 year-old Girl with Non-progressive Muscle
Weakness. May, 2003, Case 305-2. Home Page |
O. Hans Iwenofu, M.D.1, Julie T. Parke, M.D.2, and Kar-Ming Fung, M.D., Ph.D.1 Last update: May 30, 2003.
1 Department of Pathology and 2 Department of Neurology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Clinical information: The patient was a 5 year-old girl with muscle weakness and pain. Her condition was non-progressive. She had chronic pain in the leg since 18 months of age and the pain tended to occur at around 5 o'clock in the afternoon. There was no cramping. Her motor milestones were slightly delayed; as per her mother, she started walking and running later than other children but the exact delay was not clear. She had mild difficulty in climbing stairs and needed to use hand rails. She also had pelvic girdle weakness and some truncal hypotonia and instability. A partial Gower's maneuver was necessary when she arose from a supine position. There was no evidence of decrease in facial weakness or facial expression. There was an absent of joint reflex. Her serum creatine kinase level was normal. Her mother also suffered some muscle pain as a child and had decreased facial expression. A muscle biopsy from the left thigh muscle was performed and yielded the following specimen.
Pathology of the case:
Abbreviations:
| HE | Hematoxylin-eosin stain. | COX | Cytochrome C oxidase reaction. |
| MGT | Modified Gomori's trichrome stain. | PAS | Periodic acid Schiff reaction. |
| NADH-TR | NADH-tetrazolium reductase reaction. | Desmin | Immunohistochemistry for desmin. |
| SDH | Succinate dehydrogenase reaction. | EM | Electron microscopy |
Immunohistochemistry was performed on formalin fixed, paraffin embedded tissue. Histochemical reactions were performed on frozen tissue.
|
|
|
|
|
|
|
| A. |
B. ATPase pH 9.6 |
C. Trichrome |
D. NADH-TR |
E. SDH |
F. COX |
|
|
|
|
|||
|
G. PAS |
H. Desmin |
I. Electron miroscopy |
On hematoxylin-eosin stained frozen sections, there was mild variation in fiber diameter. A vaguely defined centrally located round to oval core of granular and slightly refractile area could be seen in manay fibers (Panel A) but these areas were very subtle. These areas remained very subtle in modified Gomori's trichrome but they are slightly more impressive than those in the hematoxylin-eosin stained sections (Panel B). An ATPase reaction performed at pH 9.4 disclosed predominantly type 1 fibers (Panel C). This finding was confirmed by ATPase reaction performed at pH 4.3 and 4.6. Many of the fibers had a centrally locater round to oval areas that were devoid of NADH-TR histochemical reactivity (Panel D). Similar features were demonstrated by SDH and COX (Panel E and F). The cores were not readily demonstrated by PAS stain (Panel G). On paraffin sections, the cores were well demonstrated by immunohistochemistry for desmin (Panel H). At ultrastructural level, the cores were well circumscribed volumnes of myofibrils that were in different register with the surrounding normal appearing myofibrils (Panel I). There was reduction in length of I-bands, Z-disc streaming and dissolution of Z-disc (Panel J).
|
DIAGNOSIS: Central core disease. |
Discussion: General Information Genetics Pathology
General Information
Central core disease (CCD) is one of the first congenital myopathies
recognized and was initially described by Shy and Magee in 1956
1
from a series of five patients, one female and four males in three generations
of the same family. CCD is a non-progressive myopathy that
often has its first manifestation in childhood. The muscle weakness is usually
mild to moderate and is compatible with long-term survival. Most of the cases
are transmitted as autosomal dominant trait. The pathologic hallmark is a
centrally located core in a muscle fiber that is composed of disorganized
myofibrils 2,3,4.
Patients are susceptible to developing malignant hyperthermia.
Mutations in the gene for the ryanodine receptor (RYR 1) on
chromosome 19q, the gene related to malignant hyperthermia
5,
have been found in some patients.
Although CCD typically manifests at or shortly after birth, initial presentation
has been documented also in adults. For
the typical cases, the patients develop mild and non-progressive muscle weakness
during infancy or childhood and have delayed development of motor milestones.
The weakness is usually mild to moderate and is compatible with long-term
survival. While some patients may remain asymptomatic, some patients may have
significant disability and are wheelchair bound.. The weakness is either
proximal or generalized. Severe infantile hypotonia, however, is not a typical
feature. Muscles
are thin and tendon reflexes are often preserved.
The patients often have difficulties in climbing stairs and running. Rising from
a supine position may be problematic enough to require Gower's
maneuver. Facial weakness is rather typical for CCD but is, like other signs and
symptoms, not always present.
Many patients are unable to totally bury their eyelashes. Patients may have
painless or almost painless muscle cramps after exercise. The serum creatine
kinase is sometimes elevated. Congenital dislocation hip dislocation, pes cavus,
and kyphoscoliosis are common. As a result, patients may have bizarre posture at
the time of presentation.
CCD is most often an autosomal dominant disorder. Some sporadic cases and putative
autosomal recessive inheritance have been described
6.
The gene related to CCD has been mapped to chromosome 19q12-13.2 and its RYR
1 region
7,
8,
9.
Ryanodine
receptor contains the channels for calcium release from the sarcoplasmic
reticulum. Mutations in ryanodine receptor (RYR 1) gene on chromosome
19q12-13.2 have been found in patients with malignant hyperthermia.
Interestingly, many patients with CCD are
susceptible to develop malignant hyperthermia.
The
caffeine-halothane contracture test (CHCT) is the only recognized laboratory
test to diagnose malignant hyperthermia
10.
CCD
and malignant hyperthermia may be different expressions of mutations of the same
gene. Patients with CCD may represent a subgroup of patients and families within
the much larger family of malignant hyperthermia. The relationship of RYR 1 gene mutation and pathogenesis of CCD
has not been established. The weakness in central core disease is probably
related to abnormal calcium channel functions due to mutation of ryanodine
receptor
11.
Immunohistochemical
demonstration of an increased ryanodine
receptor within
the cores suggests a pathogenetic role in the formation of cores by ryanodine
receptor
12.
The histopathologic hallmark of CCD is the formation of cores. The cores are
large round areas that occupy about 30-60% of the cross sectional surface of
muscle fibers, they may be central or eccentric. Type 1 fibers are more often
affected and the proportion of fibers being affected is quite variable. They
tend to increase in number with age. Cores are not readily seen with hematoxylin
and eosin stain or modified Gomor’s trichrome stain but they stand out
brilliantly as round to oval areas devoid of oxidative enzyme activities. The
cores tend to be separated by a slim ring of increased oxidative activity and
sometimes also by lipid droplets. Cores also lack periodic acid schiff (PAS)
reactivity. Typically, the cores run along the long axis of the muscle fibres
across numerous sarcomeres. A longitudinal section is very helpful in
identifying this feature. Immunoreactivity
of desmin is reduced or, in contrast, appear as strongly positive spots within the cores.
Increased expression of desmin is seen in the non-core zones.
Immunohistochemistry is a good way to demonstrate the length of the cores in
longitudinal sections since they are more readily available in paraffin
sections.
Central cores should be distinguished from target fibers. Similar to cores, the center of target fibers contains myofibrils with various forms of dissolution and disorganization and most target fibers are type 1 fibers. However, targets have a three zone architecture but cores often have only a two zone architecture on histochemistry for oxidative enzymes. The most reliable distinction at the level of light microscopy is their length. The length of targets are limited and do not extend across more than a few sarcomeres (up to 500 mm). Cores, in contrast, may run the entire length of the fiber.
 Click thumbnail to see target fibers for comparison.
Click thumbnail to see target fibers for comparison.
Central cores are not entirely specific for CCD. Both cores and nemaline rods
have been demonstrated in a family with Thr4637Ala
mutation in the transmembrane region of the ryanodine receptor protein
12.
They
have also been described in a soleus muscle biopsy of patient with familial
hypertrophic cardiomyopathy due to a mutation in the beta myosin heavy chain
gene (MYH 7) suggesting genetic heterogeneity of CCD
13.
Ultrastructurally, a core is a volume of well circumscribed myofibrils that have either a different register of sarcomeres or even disorganized sarcomeres surrounded by normal myofibrils with normal appearing architecture. The sarcomeres are usually contracted leading to decreased length of the I-band. The Z disc is less straight. Mitochondria are rare in the core with a tendency to accumulate at the interphase between the normal myofibrils and the cores. Glycogen particles are less numerous in the core than elsewhere. and triads may be present normally or distorted. Z-disc streaming, when present in the core, is associated with loss and disarrangement of myofilaments and disturbance of sarcotubular structure. Neville and Brooke 14 recognized two types of cores: structured and unstructured. Structured cores show increased myofibrillar ATPase activity while the activity is decreased in unstructured cores. Ultrastrucurally the unstructured cores show considerable disorganization, staggering of Z discs, elongation of T-tubules, disorientation of triads and frequently Z-disc streaming. In our experience, cores often have morphologic features in a hybrid form of structured and unstructured cores as described by Neville and Brooke.
Reference:
Shy
GM, Magee KR. A new congenital non-progressive myopathy. Brain
1956 79:610-9.
Goebel
HH. Central core disease. In Karpati G (ed.): Structural
and Molecular Basis of Skeletal Muscle Diseases.
World Federation of Neurology, Basel, 2002, pp. 65-67.
Carpenter
S and Karpati G. Pathology
of skeletal muscle.
Oxford University Press, New York, 2001, 2nd edition, pp. 65-67.
Dubowitz V. Muscle Disorders in Childhood. WB Saunders, Philadelphia, 1995, 2nd edition, pp.135-142.
Brandt
A, Schleithoff L, Jurkat-Rott K, Klingler W, Baur C, Lehmann-Horn F.
Screening of the ryanodine receptor gene in 105 malignant
hyperthermia families: novel mutations and concordance with the in vitro
contracture test. Hum Mol Genet. 1999 8:2055-62.
Manzur
AY, Sewry CA, Ziprin J, Dubowitz V, Muntoni F. A
severe clinical and pathological variant of central core disease with
possible autosomal recessive inheritance. Neuromuscul Disord. 1998 8:467-73.
Haan
EA, Freemantle CJ, McCure JA, Friend KL, Mulley JC. Assignment
of the gene for central core disease to chromosome 19. Hum Genet.
1990 86:187-90.
Kausch
K, Lehmann-Horn F, Janka M, Wieringa B, Grimm T, Muller CR. Evidence
for linkage of the central core disease locus to the proximal long arm of
human chromosome 19. Genomics. 1991 10:765-9.
Mulley
JC, Kozman HM, Phillips HA, Gedeon AK, McCure JA, Iles DE, Gregg RG, Hogan
K, Couch FJ, MacLennan DH, et al. Refined
genetic localization for central core disease.
Am J Hum Genet. 1993 Feb;52(2):398-405.
Allen
GC, Larach MG, Kunselman AR.
The sensitivity and specificity of the caffeine-halothane contracture test:
a report from the North American Malignant Hyperthermia Registry. The North
American Malignant Hyperthermia Registry of MHAUS.
Anesthesiology. 1998 88:579-88.
Dirksen
RT, Avila G. Altered
ryanodine receptor function in central core disease: leaky or uncoupled
Ca(2+) release channels? Trends Cardiovasc Med. 2002 12:189-97.
Scacheri PC, Hoffman EP, Fratkin JD, Semino-Mora C, Senchak A, Davis MR, Laing NG, Vedanarayanan V, Subramony SH. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000 55:1689-96.
Fananapazir
L, Dalakas MC, Cyran F, Cohn G, Epstein ND. Missense
mutations in the beta-myosin heavy-chain gene cause central core disease in
hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 1993
90:3993-7.