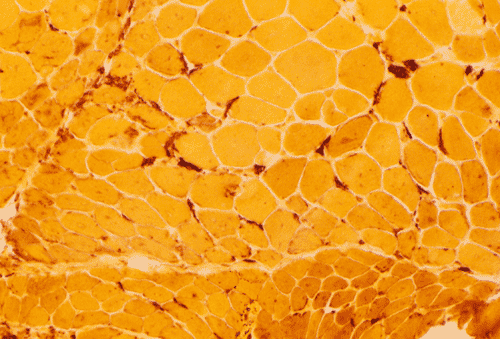
Trichrome
ATPase pH 9.6
Esterase

| A 56 year-old Woman with
a Rash and Generalized Weakness. October, 2003, Case 310-1. Home Page |
Chimène Kesserwan, M.D.1, Hamid Sami, M.D. 2, Zahid F. Cheema, M.D. 2, Kar-Ming Fung, M.D., Ph.D. 1 Last Update September 30, 2003.
1 Department of Pathology and 2 Department of Neurology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Clinical information:
General
The patient is a 56-year old right-handed woman with hypothyroidism, depression and kidney stone. She developed rash on her chest a year ago and that has progressed to involve her face, arms and trunk. She reported pain and stiffness in her hands and joints and developed gradual weakness in her arms and legs that has recently progressed to the severity that limit her activities in daily living and chores. She was started on Prednisone with some improvement in her symptoms but when she was tapered down on Prednisone, her symptoms would resume. She does not report any breathing problem, swallowing problem, weight loss, night sweats or any bowel or bladder incontinence.
Neurologic examination
MS and Cranial nerves: Normal cortical function; normal cranial nerve function.
Motor: neck strength was normal. In the upper extremities, she has proximal weakness with 4+/5 in deltoid, biceps and triceps; distal strength is 5/5. In the lower extremities, there is proximal weakness with 4/5 strength in gluteus medius and normal strength distally. There is no evidence of wasting or reduced muscle tone.
Sensory: Intact to pinprick, temperature, vibration and position.
Reflex: Absence of ankle reflexes, 1/5 in all the all other areas, no Babinski sign.
Gait/Coordination: Positive Gower's sign, normal gait, slightly difficulty in tendem walking.
Laboratory studies
Erythrocyte sedimentation rate (ESR): 57 mm/hr (normal is 0-20 mm/hr).
Serum creatine kinase (CPK): 278 U/L (normal is 20-140 U/L).
Nerve conduction studies/EMG: Moderately severe, nonspecific, generalized myopathy with electrophysiologic evidence of short duration and amplitude of motor potentials without active denervation or reinnervation in her proximal arm and leg muscles.
Diagnostic procedure
A biopsy was performed. The followings are representative photomicrographs of the biopsy material.
|
|
|
|
|
|
|
| A. | B. |
C. Trichrome |
D. ATPase pH 9.6 |
E. Esterase |
F. |
|
|
|
|
|
||
| G. | H. | I. | J. |
Pathology of the case:
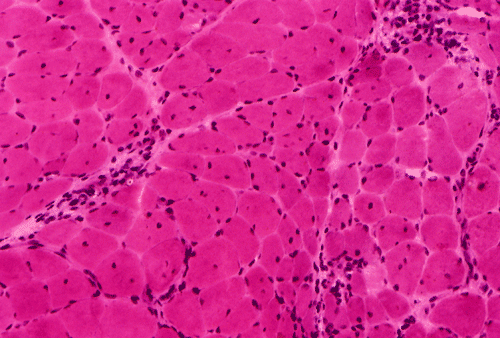
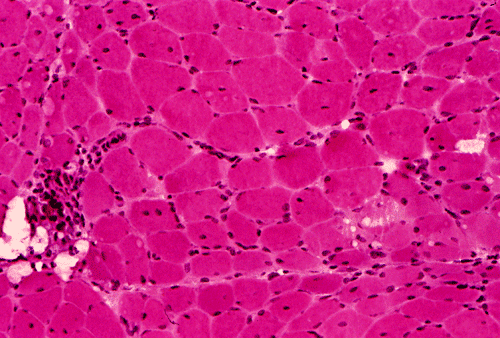
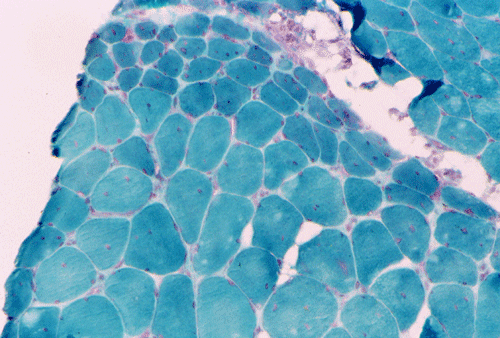
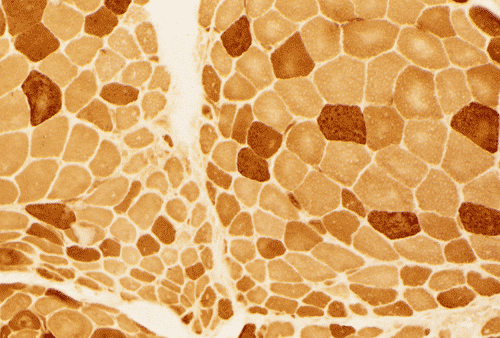
Light microscopy and histochemistry: Chronic inflammatory cell infiltration is well identified in the hematoxylin-eosin stained frozen section. The bulk of these inflammatory cells are angiocentric and with interstitial extension, i.e., in between muscle fibers, with occasional inflammatory cells infiltrating into the muscle fiber (Panel A and B). Necrotic fibers infiltrated by histiocytes are often common and they are best evaluated with paraffin sections. Most of the fibers appear more round to oval than polygonal and there is an increase in centrally located nuclei. These are both signs of chronicity. Under normal circumstances, there should not be more than 3 to 5 centrally located nuclei. There is also variation in fiber diameter. Although no hypertrophic fibers are seen, the smaller fibers being illustrated are atrophic fibers. Modified Gomori's trichrome (Panel C) is a stain that is very helpful in identification of intracytoplasmic inclusions such as nemaline bodies, subsarcolemmal depositions and ragged red fibers. In this case, there are no intracytoplasmic inclusions or subsarcolemmal deposition. However, this photo is taken at the edge of the specimen and disclosed the tendency for the small fibers to be be found around the muscle fascicles, this phenomenon is called perifascicular atrophy and is commonly seen in dermatomyositis. In the ATPase reaction performed at pH 9.4, the type I (pale) and type II (dark) fibers can be well recognized. There is no evidence of fiber type grouping (groups of small fibers of the same type, often a sign of neurogenic atrophy). Inflammatory cells infiltrating the muscle are stain dark with esterase reaction.
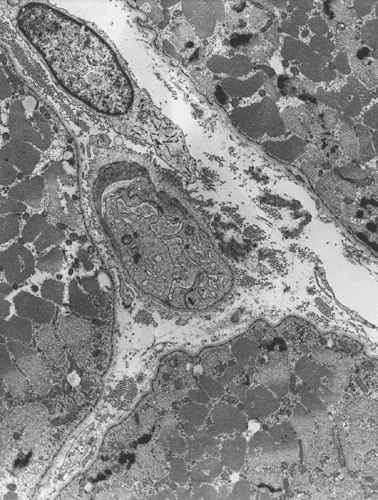
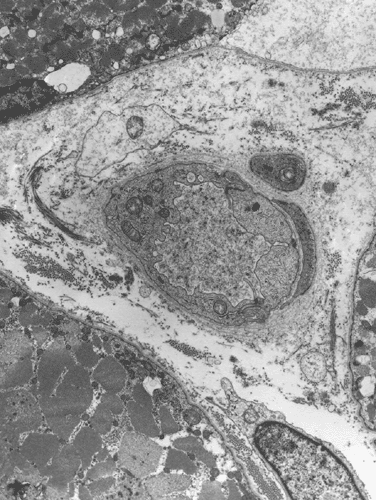
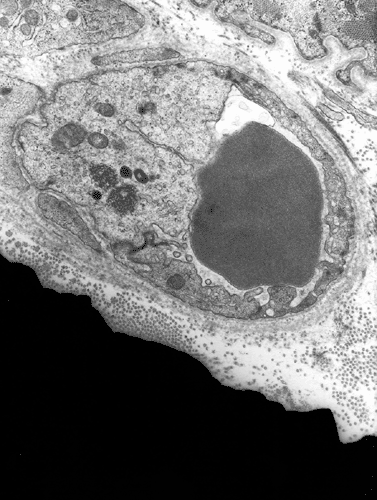
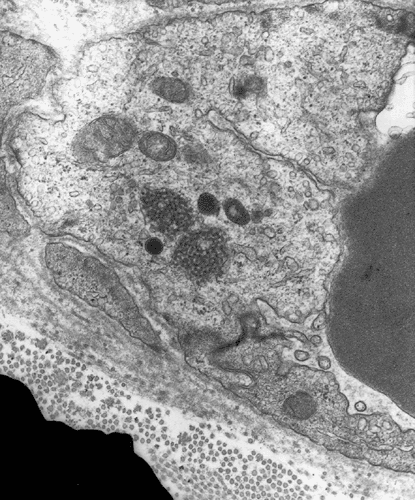
Electron microscopy: The inflammatory cells and necrotic muscle fibers are not shown here. The highlight of this case is endotherlial changes. In many of the capillaries, the endothelial cells have enlarged cytoplasm with reduction of luminal diameter (Panel F and G). Sometimes necrotic debris are present in the lumen (not illustrated in this case). On close examination of the endothelial cytoplasm, collections of tubular reticular structures are found (Panel H and I). These are common findings in dermatomyositis. In some fibers, z-disc streaming are identified (Panel J).
|
DIAGNOSIS: Dermatomyositis |
Comment: Dermatophysitis is essentially a clinicopathologic diagnosis. The clinical and pathologic features in this case are consistent with dermatophysitis. Perifascicular atrophy, when present, is strongly suggestive of dermatomyositis. They must be distinguished from neurogenic type of atrophy that is often seen in inclusion body myositis. ATPase preparation is often helpful. Endothelial damage is seen in most cases of dermatomyositis. In addition to enlarged endothelial cells and tubular reticular structures, dense bodies, undulating tubules in endoplasmic reticulum and cylindrical confronting cisternae are common features. The later three features are not seen in this case. Z-disc streaming is a resulted from disorganized z-disc. It typically starts and stops at the z-disc. It is a common and non-specific change that can be encountered in many myopathies. Z-disc streaming has also been described in normal control subjects.
Discussion: General Information Laboratory findings Pathology Differential diagnosis
General Information
Dermatomyoitis belongs to the family of idiopathic inflammatory myopathies which
encompass a heterogenous group of chronic inflammatory disorders of skeletal
muscle. Polymyositis and inclusion body myositis are the two other major
categories of idiopathic inflammatory myopathy. Their etiology is not clear. The
pathologic change is characterized by chronic inflammatory cell infiltration and
perifascicular atrophy.
Dermatomyositis is common and comprises over one-third of all adult cases of
inflammatory myopathies and almost all juvenile (childhood) inflammatory
myopathy. It has
different clinical features in adult and children but similar pathology. In
adults, the peak incidence occurs between 40-70 years of age. Women are affect
two to three times more often than male. In children, the peak is around 10
years of age.
Dermatomyositis is a systemic disease with major manifestation in the skin and
skeletal muscle. As compared with the normal population, adults with
dermatomyositis have a higher risk of developing visceral cancers. About 20% of
the adult patients, particularly the older patients harbour an internal
malignancy. There is no such correlation in juvenile cases. The cardinal triad
includes skin rash, muscle weakness and constitutional symptoms such as malaise,
listlessness and lethargy. Low-grade fever is common. Joint pain,
arthritis, dysphagia and Reynaud’s phenomenon are common. Colonic
perforation due to vascular occlusion, although uncommon, has been documented. Lymphadenopathy
and splenomegaly can occur. The clinical signs and symptoms are similar to that
of connective tissue diseases. Respiratory,
cardiovascular, gastrointestinal, and renal functions may all be altered.
The clinical manifestations of dermatomositis in adults vary significantly. The
typical picture is featured by subacute onset of skin rash, and muscle pain and
weakness. The clinical manifestations may evolve over a period of 2 to 3 weeks.
Skin rash is evident in most patients and usually precedes the muscular
manifestation. The typical cutaneous features include erythema over the light
exposed skin including the cheeks, the bridge of the nose, upper anterior chest,
upper posterior chest, and knuckles. There may be a scaly eruption (Grottron’s
sign) over the knuckles but not the phalanges. Some patients may develop “mechanic’s
hands” (i.e., dry, crackled hands Patients also have photosensitivity.
Periorbital and perioral swelling may occur, particularly in fulminant cases.
Muscle weakness is slow in onset, bilateral, symmetrical, and typically affects the proximal muscle first. The muscle pain can be worsened by exercise. Electromyography shows a myopathic pattern of motor unit potential and spontaneous “irritative” activity (fibrillation potentials and positive sharp waves). Extramuscular manifestations, including interstitial lung disease, vasculitis and myocarditis, may be present in some cases. Calcinosis is much more common in juvenile than adult cases, usually in the intertistitial tissue of the muscle or subcutaneous tissue. Calcification is most often found two years after the onset of disease and is a good indicator of chronicity. Some calcifications may subsequently resolve. Palpable nodules may be present, especially in juvenile cases and contracture is common.
Serum creatine kinase is often, but not always, elevated and typically to a moderate level, and sometimes, high level. Rhabdomyolysis may develop in some patients and lead to myoglobinuria and a transient and extremely high serum creatine kinase level.A variety of autoantibodies have been identified in inflammatory myopathies. Some of these are myositis-specific (i.e., occurring only in pathes with inflammatory myopathies), the others are myositis-associated (i.e., found in patients with inflammatory myopathy and other connective tissue tiseases). The discovery of autoantibodies strongly points to an autoimmune mechism. These antibodies, however, are only of limited value in diagnosis and management. Among these autoantibodies, anti-Mi-2 antibodies are present in high titer in up to 35% of dermatomyositis including some juvenile cases. Anti-Jo-1 antibodies are associated with antisynthetase syndrome featured by myositis, interstitial lung disease, arthritis, Raynaud’s phenomenon, and “mechanic’s hands”. Other autoantibodies are far less specific for diagnostic purpose.
The pathology varies from case to case and from muscle to muscle. In general,
there is chronic inflammatory cell infiltration associated with necrotic fibers
and regenerating fibers. The inflammatory cell infiltrations consist of
lymphocytes, macrophages, and plasma cells with antigenctric arrangement with
extension into the septae and, less commonly into fascicles.
There is increased variation in fiber diameter and rounding of muscle fibers.
Atrophic fibers are present and both type I and II fibers are involved. The
atrophic fibers tend to occur at the perimeter of the fascicle, the so-called
“perifascicular atrophy”. The atrophic fiberes are often darker stained than
type I fibers with NADH-TR reaction. Perifascicular atrophy is, in fact, best
detected by NADH-TR reaction. Unless there is a co-existing neurogenic atrophy,
fiber type grouping or other evidence of neurogenic atrophy should not be
present. Necrotic fibers are seen in most adult cases and often in the form of
single necrotic fiber at the edge of the fascicles. Bundles of necrotic fibers
at the center of the fascicles suggesting infarction may be present. Macrophages
may not be abundant and the necrotic foci are surrounded by plump myoblasts or
myotubules. Inflammatory cells are strongly positive for esterase reaction.
Unlike polymyositis, partial invasion of non-necrotic muscle fibers is not seen.
Many featureless necrotic fibers not infiltrated by chronic inflammatory cells
are present, the so-called “delayed phagocytosis”. Necrotic fibers are less
common in juvenile cases. In contrast, juvenile cases tend to have punched-out
areas (vacuoles) of myofibrillar loss within fibers. On modified Gomori’s
trichrome, z-disc streaming in the form of poorly defined patchy dark staining
can be demonstrated.
Regenerating fibers are usually found in the same distrubution as necrotic
fibers. They may appear as groups of small fibers. Similar to necrotic fibers,
they are more commonly seen in adult cases than in juvenile cases. There is a
progressive destruction of capillaries with reduced number of capillaries. This
feature varies from region to region of the biopsy material. Thrombosed and
recanalized vessels can occur.
Immunohistochemically, Both B-cells and T-cells are present. While the number of
B-cells and T-cells are similar in infiltration around blood vessels, B-cells
are a lot less common in the infiltrations within fascicles. CD4 cells
predominate. The CD4/CD8 ratio is highest at perivascular infiltrates and lowest
in endomysial infiltrates. Class I MHC antigens are expressed by damaged fibers but, unlike polymyositis,
not usually in intact fibers. Membrane
attack complex in capillary
walls can be demonstratrated by immunohistochemistry in both muscle and skin
lesions. Acid phosphatase is demonstrated in blood vessels indicating
phagocytosis of capillary debris.
Differential diagnosis
The diagnosis of inflammatory
myopathy is a clinicopathologic diagnosis. Evidence from both the clinical and
laboratory sides should be considered. The major differential diagnoses are
polymyositis and inclusion body myositis.
Polymyositis usually evolves over several months and is slower than dermatomyositis (usually over weeks) and but faster than inclusion body myositis. Polymyositis is not associated with cutaneous manifestions of dermatomyositis and practically do not occur in children. There is also histologic differences and are best illustrated in a table form. [Click here to see the table]
Inclusion body myositis is the most common
acquired myopathy in patients over 50 years of age and is more common in man.
Unlike polymyositis and dermatomyositis, inclusion body myositis is not
responsive to steroid Histologically, the
characteristic features include rimmed vacuoles and atrophic muscle bundles with
a pattern that resembles neurogenic atrophy.
There are many clinical and pathologic similarities between systemic lupus
erythematosus (SLE) and dermatomyositis. In fact, a myositis that is
indistinguishable from dermatomyositis on biopsy material is knows to patients
with SLE. Distinction between the two entities depends on clinical and
laboratory findings.
Further Reading:
Levine
TD.
History of dermatomyositis. Arch
Neurol. 2003 May;60(5):780-2. No abstract available.
Younger
DS.
The myopathies. Med
Clin North Am. 2003;87:899-907, ix.
Hilton-Jones
D.
Inflammatory muscle diseases. Curr
Opin Neurol. 2001;14:591-6.
Hilton-Jones
D.
Diagnosis and treatment of inflammatory muscle diseases. J
Neurol Neurosurg Psychiatry. 2003;74 Suppl 2:ii25-ii31.
Mastaglia
FL, Garlepp MJ, Phillips BA, Zilko PJ.
Inflammatory
myopathies: clinical, diagnostic and therapeutic aspects. Muscle
Nerve. 2003;27:407-25.
Dalakas
MC.
Muscle
biopsy findings in inflammatory myopathies. Rheum
Dis Clin North Am. 2002;28:779-98, vi.
Dorph
C, Lundberg IE.
Idiopathic inflammatory myopathies - myositis. Best
Pract Res Clin Rheumatol. 2002;16:817-32.
van
Paassen P, Damoiseaux J, Cohen Tervaert JW.
Laboratory
assessment in musculoskeletal disorders. Best
Pract Res Clin Rheumatol. 2003;17:475-94.
Greenberg
SA, Sanoudou D, Haslett JN, Kohane IS, Kunkel LM, Beggs AH, Amato AA.
Molecular profiles of inflammatory myopathies. Neurology.
2002;59:1170-82.
Targoff
IN.
Idiopathic
inflammatory myopathy: autoantibody update. Curr
Rheumatol Rep. 2002;4:434-41.
Lim
Y, Lee DY, Lee S, Park SY, Kim J, Cho B, Lee H, Kim HY, Lee E, Song YW,
Jeoung DI.
Identification
of autoantibodies associated with systemic lupus erythematosus. Biochem
Biophys Res Commun. 2002;295:119-24.
Kashiwabara
K, Ota K.
Rapidly progressive interstitial lung disease in a dermatomyositis patient
with high levels of creatine phosphokinase, severe muscle symptoms and
positive anti-Jo-1 antibody. Intern Med. 2002;41:584-8.
Arnett
FC, Hirsch TJ, Bias WB, Nishikai M, Reichlin M.
The Jo-1 antibody system in myositis: relationships to clinical features and
HLA. J Rheumatol. 1981;8:925-30.